
Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
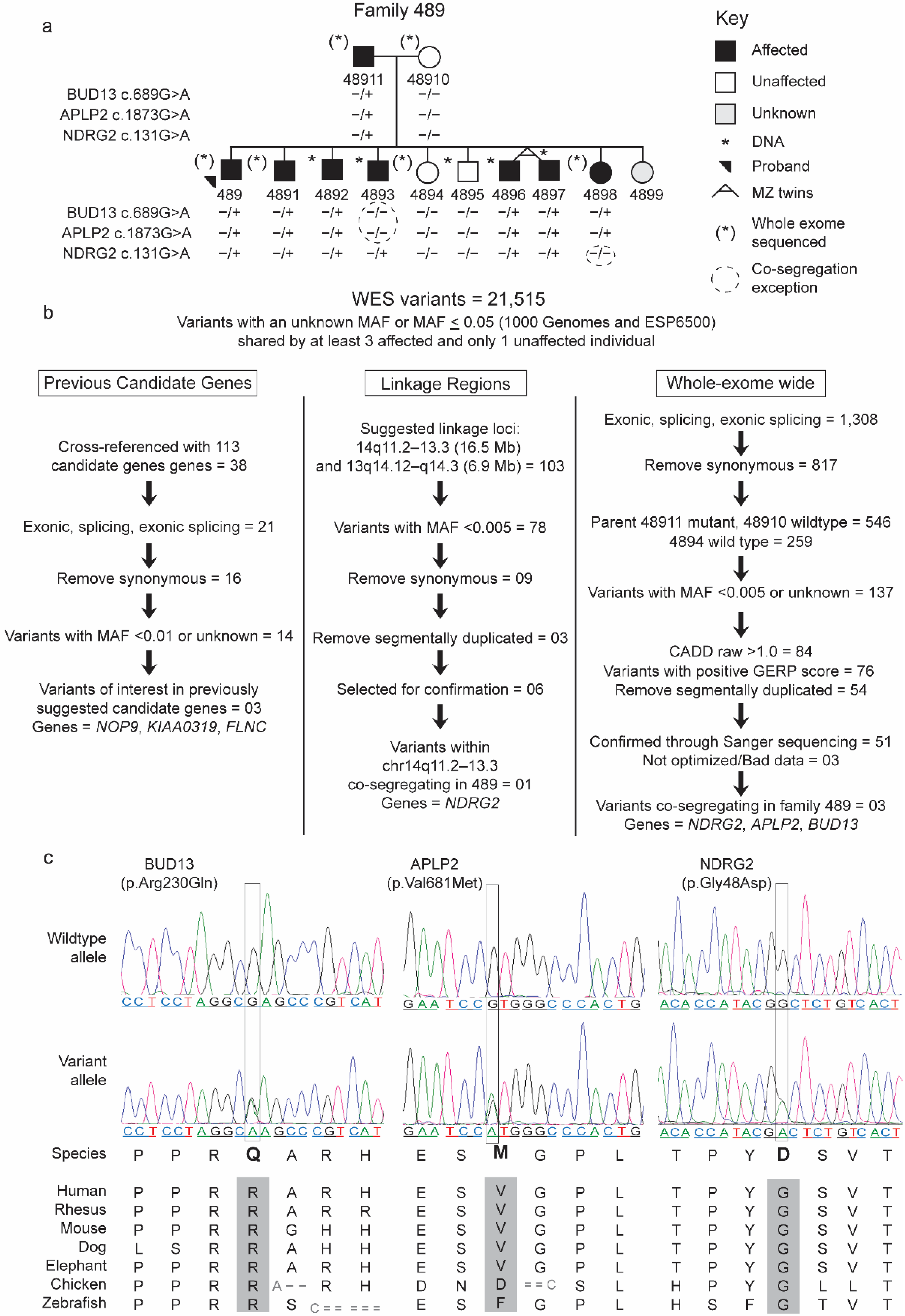
2.1.1. Family 489
2.1.2. Additional Participants
2.1.3. Phenotype
2.2. Genetic Analyses
2.2.1. DNA Collection and Preparation
2.2.2. Whole-Exome Sequencing and Data Analysis
2.2.3. Prioritization of Rare Variants in the WES
2.2.4. Identification of Candidate Genes, Confirmation, and Significance Testing

{kind=link}
{kind=link}
| Fam 489 Variants | Additional Variants | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Previous Candidates | Co-Segregating | NDRG2 | APLP2 | BUD13 | ||||||||||||||||
| Evidence Class | Evidence | KIAA0319 rs113411083 | FLNC rs202223616 | NOP9 rs183868211 | NDRG2 rs11552412 | APLP2 rs370970986 | BUD13 rs139478949 | rs779725845 | rs1063201 | chr11:129992279 | rs201861910 | rs35585096 | rs116087150 | rs1467808735 | rs144776650 | rs11216131 | rs1427011653 | rs145410701 | rs61730763 | rs145906707 |
| Genetic | MAF ≤ 0.05 | + | + | − | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Co-segregation | − | − | − | + | + | + | − | − | − | + | − | − | − | − | − | − | − | − | + | |
| ≥1 proband | − | − | + | − | − | − | − | − | − | − | + | − | − | + | − | − | − | − | + | |
| Informatic | Positive GERP Score | + | + | + | + | + | + | + | − | + | − | + | + | + | − | + | − | − | + | + |
| Total # of damaging in silico scores | 2 | 1 | 0 | 4 | 2 | 4 | 2 | 0 | 2 | NA | 2 | 4 | 4 | 1 | 5 | 0 | 0 | 3 | NA | |
| HOPE output/AA change | ||||||||||||||||||||
| Size | ∧ | ∧ | ∨ | ∧ | ∧ | ∨ | ∧ | ∧ | ∧ | NA | ∧ | ∨ | ∨ | ∨ | ∨ | ∧ | ∨ | ∧ | NA | |
| >Hydrophobic | + | − | + | + | − | − | + | − | − | NA | + | + | + | − | + | − | + | − | NA | |
| Charge change | pos | neg | pos | neu | NC | pos | NC | NC | neg | NA | NC | pos | neu | pos | pos | NC | NC | NC | NA | |
| to | to | to | to | to | to | to | to | to | to | |||||||||||
| neu | pos | neu | neg | neu | pos | neu | neg | neu | neu | |||||||||||
| Causality | P | B | B | P | P | P | P | B | P | NA | P | P | P | B | P | B | B | P | NA | |
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomblin, J.B.; Records, N.L.; Buckwalter, P.; Zhang, X.; Smith, E.; O’Brien, M. Prevalence of Specific Language Impairment in Kindergarten Children. J. Speech Lang. Hear. Res. 1997, 40, 1245–1260. [Google Scholar] [CrossRef]
- Norbury, C.F.; Gooch, D.; Wray, C.; Baird, G.; Charman, T.; Simonoff, E.; Vamvakas, G.; Pickles, A. The impact of nonverbal ability on prevalence and clinical presentation of language disorder: Evidence from a population study. J. Child Psychol. Psychiatry 2016, 57, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Deafness and Other Communication Disorders. Specific Language Impairment. Available online: https://www.nidcd.nih.gov/health/specific-language-impairment (accessed on 10 November 2017).
- Rice, M.L. Overlooked by Public Health: Specific Language Impairment. Open Access Government. 2017. Available online: https://www.openaccessgovernment.org/overlooked-public-healthspecific-language-impairment/34474/ (accessed on 26 October 2021).
- Brownlie, E.B.; Graham, E.; Bao, L.; Koyama, E.; Beitchman, J.H. Language disorder and retrospectively reported sexual abuse of girls: Severity and disclosure. J. Child Psychol. Psychiatry 2017, 58, 1114–1121. [Google Scholar] [CrossRef]
- Conti-Ramsden, G.; Botting, N. Social Difficulties and Victimization in Children With SLI at 11 Years of Age. J. Speech Lang. Hear. Res. 2004, 47, 145–161. [Google Scholar] [CrossRef]
- Bishop, D.V.M.; North, T.; Donlan, C. Genetic basis of specific language impairment: Evidence from a twin study. Dev. Med. Child Neurol. 1995, 37, 56–71. [Google Scholar] [CrossRef]
- Rice, M.L.; Haney, K.R.; Wexler, K. Family Histories of Children with SLI Who Show Extended Optional Infinitives. J. Speech Lang. Hear. Res. 1998, 41, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.L.; Wexler, K. Toward Tense as a Clinical Marker of Specific Language Impairment in English-Speaking Children. J. Speech Lang. Hear. Res. 1996, 39, 1239–1257. [Google Scholar] [CrossRef]
- Rice, M.L.; Taylor, C.L.; Zubrick, S.R.; Hoffman, L.; Earnest, K.K. Heritability of Specific Language Impairment and Nonspecific Language Impairment at Ages 4 and 6 Years Across Phenotypes of Speech, Language, and Nonverbal Cognition. J. Speech Lang. Hear. Res. 2020, 63, 793–813. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.L.; Zubrick, S.R.; Taylor, C.L.; Hoffman, L.; Gayán, J. Longitudinal Study of Language and Speech of Twins at 4 and 6 Years: Twinning Effects Decrease, Zygosity Effects Disappear, and Heritability Increases. J. Speech Lang. Hear. Res. 2018, 61, 79–93. [Google Scholar] [CrossRef]
- SLI Consortium. A Genomewide Scan Identifies Two Novel Loci Involved in Specific Language Impairment. Am. J. Hum. Genet. 2002, 70, 384–398. [Google Scholar] [CrossRef]
- SLI Consortium. Highly Significant Linkage to the SLI1 Locus in an Expanded Sample of Individuals Affected by Specific Language Impairment. Am. J. Hum. Genet. 2004, 74, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Vieland, V.J.; Bassett, A.S.; Tallal, P.; Brzustowicz, L.M. A Major Susceptibility Locus for Specific Language Impairment Is Located on 13q21. Am. J. Hum. Genet. 2002, 71, 45–55. [Google Scholar] [CrossRef]
- Newbury, D.F.; Winchester, L.; Addis, L.; Paracchini, S.; Buckingham, L.-L.; Clark, A.; Cohen, W.; Cowie, H.; Dworzynski, K.; Everitt, A.; et al. CMIP and ATP2C2 Modulate Phonological Short-Term Memory in Language Impairment. Am. J. Hum. Genet. 2009, 85, 264–272. [Google Scholar] [CrossRef]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Smith, B.J.; Vieland, V.J.; Tallal, P.; Brzustowicz, L.M. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered. 2004, 57, 10–20. [Google Scholar] [CrossRef]
- Andres, E.M.; Earnest, K.K.; Smith, S.D.; Rice, M.L.; Raza, M.H. Pedigree-Based Gene Mapping Supports Previous Loci and Reveals Novel Suggestive Loci in Specific Language Impairment. J. Speech Lang. Hear. Res. 2020, 63, 4046–4061. [Google Scholar] [CrossRef]
- Nudel, R.; Simpson, N.H.; Baird, G.; O’Hare, A.; Conti-Ramsden, G.; Bolton, P.F.; Hennessy, E.R.; SLI Consortium; Ring, S.M.; Davey Smith, G.D.; et al. Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment. Genes Brain Behav. 2014, 13, 418–429. [Google Scholar] [CrossRef]
- Andres, E.M.; Hafeez, H.; Yousaf, A.; Riazuddin, S.; Rice, M.L.; Basra, M.A.R.; Raza, M.H. A genome-wide analysis in consanguineous families reveals new chromosomal loci in specific language impairment (SLI). Eur. J. Hum. Genet. 2019, 27, 1274–1285. [Google Scholar] [CrossRef]
- Truong, D.T.; Shriberg, L.D.; Smith, S.D.; Chapman, K.L.; Scheer-Cohen, A.R.; DeMille, M.M.C.; Adams, A.K.; Nato, A.Q.; Wijsman, E.M.; Eicher, J.D.; et al. Multipoint genome-wide linkage scan for nonword repetition in a multigenerational family further supports chromosome 13q as a locus for verbal trait disorders. Hum. Genet. 2016, 135, 1329–1341. [Google Scholar] [CrossRef]
- Villanueva, P.; Newbury, D.F.; Jara, L.; De Barbieri, Z.; Mirza, G.; Palomino, H.M.; Fernández, M.A.; Cazier, J.-B.; Monaco, A.P.; Palomino, H. Genome-wide analysis of genetic susceptibility to language impairment in an isolated Chilean population. Eur. J. Hum. Genet. 2011, 19, 687–695. [Google Scholar] [CrossRef]
- Villanueva, P.; Nudel, R.; Hoischen, A.; Fernández, M.A.; Simpson, N.H.; Gilissen, C.; Reader, R.H.; Jara, L.; Echeverry, M.M.; Francks, C.; et al. Exome Sequencing in an Admixed Isolated Population Indicates NFXL1 Variants Confer a Risk for Specific Language Impairment. PLoS Genet. 2015, 11, e1004925. [Google Scholar] [CrossRef]
- Villanueva, P.; De Barbieri, Z.; Palomino, H.M.; Palomino, H. High prevalence of specific language impairment in Robinson Crusoe Island. A possible founder effect. Rev. Med. Chile 2008, 136, 186–192. [Google Scholar] [PubMed]
- Nudel, R. An investigation of NFXL1, a gene implicated in a study of specific language impairment. J. Neurodev. Disord. 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Mountford, H.S.; Villanueva, P.; Fernández, M.A.; De Barbieri, Z.; Cazier, J.-B.; Newbury, D.F. Candidate gene variant effects on language disorders in Robinson Crusoe Island. Ann. Hum. Biol. 2019, 46, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Reader, R.H.; Hoischen, A.; Veltman, J.A.; Simpson, N.H.; Francks, C.; Newbury, D.F.; Fisher, S.E. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci. Rep. 2017, 7, 46105. [Google Scholar] [CrossRef] [PubMed]
- Centanni, T.M.; Green, J.R.; Iuzzini-Seigel, J.; Bartlett, C.W.; Hogan, T.P. Evidence for the multiple hits genetic theory for inherited language impairment: A case study. Front. Genet. 2015, 6, 272. [Google Scholar] [CrossRef]
- Mountford, H.S.; Newbury, D.F. The genomic landscape of language: Insights into evolution. J. Lang. Evol. 2017, 3, 49–58. [Google Scholar] [CrossRef]
- Rice, M.L.; Tager-Flusberg, H. Language Phenotypes. In Neurophenotypes; Jagaroo, V., Santangelo, S.L., Eds.; Innovations in Cognitive Neuroscience; Springer: Boston, MA, USA, 2016; pp. 227–243. [Google Scholar]
- Rice, M.L.; Hoffman, L. Predicting Vocabulary Growth in Children With and Without Specific Language Impairment: A Longitudinal Study From 2;6 to 21 Years of Age. J. Speech Lang. Hear. Res. 2015, 58, 345–359. [Google Scholar] [CrossRef]
- Rice, M.L.; Wexler, K.; Cleave, P.L. Specific Language Impairment as a Period of Extended Optional Infinitive. J. Speech Lang. Hear. Res. 1995, 38, 850–863. [Google Scholar] [CrossRef]
- Rice, M.L.; Smith, S.D.; Gayán, J. Convergent genetic linkage and associations to language, speech and reading measures in families of probands with Specific Language Impairment. J. Neurodev. Disord. 2009, 1, 264–282. [Google Scholar] [CrossRef][Green Version]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D. Wechsler Intelligence Scale for Children, 3rd ed.; The Psychological Corporation: San Antonio, TX, USA, 1991. [Google Scholar]
- Wechsler, D. Adult Intelligence Scale-Third Edition; The Psychological Corporation: San Antonio, TX, USA, 1997. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Soranzo, N. The end of the start for population sequencing. Nature 2015, 526, 52–53. [Google Scholar] [CrossRef]
- NHLBI Exome Sequencing Project (ESP) Home Page. Available online: https://evs.gs.washington.edu/EVS/ (accessed on 18 December 2020).
- Guerra, J.; Cacabelos, R. Genomics of speech and language disorders. J. Transl. Genet. Genom. 2019. [Google Scholar] [CrossRef]
- Ceroni, F.; Simpson, N.H.; Francks, C.; Baird, G.; Conti-Ramsden, G.; Clark, A.; Bolton, P.F.; Hennessy, E.R.; Donnelly, P.; Bentley, D.R.; et al. Homozygous microdeletion of exon 5 in ZNF277 in a girl with specific language impairment. Eur. J. Hum. Genet. 2014, 22, 1165–1171. [Google Scholar] [CrossRef]
- Catts, H.W.; Adlof, S.M.; Weismer, S.E. Language Deficits in Poor Comprehenders: A Case for the Simple View of Reading. J. Speech Lang. Hear. Res. 2006, 49, 278–293. [Google Scholar] [CrossRef]
- Catts, H.W.; Adlof, S.M.; Hogan, T.P.; Weismer, S.E. Are Specific Language Impairment and Dyslexia Distinct Disorders? J. Speech Lang. Hear. Res. 2005, 48, 1378–1396. [Google Scholar] [CrossRef]
- Adlof, S.M.; Hogan, T.P. If We Don’t Look, We Won’t See: Measuring Language Development to Inform Literacy Instruction. Policy Insights Behav. Brain Sci. 2019, 6, 210–217. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.R.; Nelson, S.F. Calculating the statistical significance of rare variants causal for Mendelian and complex disorders. BMC Med. Genom. 2018, 11, 53. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Sunyaev, S.R.; Eisenhaber, F.; Rodchenkov, I.V.; Eisenhaber, B.; Tumanyan, V.G.; Kuznetsov, E.N. PSIC: Profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. Des. Sel. 1999, 12, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Venselaar, H.; Beek, T.A.H.T.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Neale, M.C.; Cardon, L.R. Methodology for Genetic Studies of Twins and Families; Springer Science & Business Media: Dordect, South Holland, The Netherlands, 2013. [Google Scholar]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; Van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2016, 22, 1604–1614. [Google Scholar] [CrossRef]
- Peter, B.; Wijsman, E.M.; Nato, A.Q., Jr.; University of Washington Center for Mendelian, G.; Matsushita, M.M.; Chapman, K.L.; Stanaway, I.B.; Wolff, J.; Oda, K.; Gabo, V.B.; et al. Genetic Candidate Variants in Two Multigenerational Families with Childhood Apraxia of Speech. PLoS ONE 2016, 11, e0153864. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, K.A.; Frinton, E.; Nudel, R.; Chan, M.T.M.; Thompson, P.; Hayiou-Thomas, M.E.; Talcott, J.B.; Stein, J.; Monaco, A.P.; Hulme, C.; et al. Further evidence for a parent-of-origin effect at the NOP9 locus on language-related phenotypes. J. Neurodev. Disord. 2016, 8, 24. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic Inheritance in Bardet-Biedl Syndrome, a Mendelian Recessive Disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef]
- Eichers, E.R.; Lewis, R.A.; Katsanis, N.; Lupski, J.R. Triallelic inheritance: A bridge between Mendelian and multifactorial traits. Ann. Med. 2004, 36, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Katsanis, N. Genetic Modifiers and Oligogenic Inheritance. Cold Spring Harb. Perspect. Med. 2015, 5, a017145. [Google Scholar] [CrossRef]
- Devanna, P.; Chen, X.S.; Ho, J.; Gajewski, D.; Smith, S.D.; Gialluisi, A.; Francks, C.; Fisher, S.E.; Newbury, D.F.; Vernes, S.C. Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders. Mol. Psychiatry 2017, 23, 1375–1384. [Google Scholar] [CrossRef]
- Devanna, P.; Van De Vorst, M.; Pfundt, R.; Gilissen, C.; Vernes, S.C. Genome-wide investigation of an ID cohort reveals de novo 3′UTR variants affecting gene expression. Hum. Genet. 2018, 137, 717–721. [Google Scholar] [CrossRef]
- Enard, W.; Przeworski, M.; Fisher, S.E.; Lai, C.S.; Wiebe, V.; Kitano, T.; Monaco, A.P.; Paabo, S. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 2002, 418, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.S.L.; Fisher, S.E.; Hurst, J.A.; Vargha-Khadem, F.; Monaco, A.P. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 2001, 413, 519–523. [Google Scholar] [CrossRef]
- Raza, M.H.; Mattera, R.; Morell, R.; Sainz, E.; Rahn, R.; Gutierrez, J.; Paris, E.; Root, J.; Solomon, B.; Brewer, C.; et al. Association between Rare Variants in AP4E1, a Component of Intracellular Trafficking, and Persistent Stuttering. Am. J. Hum. Genet. 2015, 97, 715–725. [Google Scholar] [CrossRef]
- Kazemi, N.; Estiar, M.A.; Fazilaty, H.; Sakhinia, E. Variants in GNPTAB, GNPTG and NAGPA genes are associated with stutterers. Gene 2018, 647, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Domingues, C.E.F.; Webster, R.; Sainz, E.; Paris, E.; Rahn, R.; Gutierrez, J.; Chow, H.M.; Mundorff, J.; Kang, C.-S.; et al. Mucolipidosis types II and III and non-syndromic stuttering are associated with different variants in the same genes. Eur. J. Hum. Genet. 2016, 24, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Drayna, D. A role for inherited metabolic deficits in persistent developmental stuttering. Mol. Genet. Metab. 2012, 107, 276–280. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reader, R.H.; Covill, L.E.; Nudel, R.; Newbury, D.F. Genome-Wide Studies of Specific Language Impairment. Curr. Behav. Neurosci. Rep. 2014, 1, 242–250. [Google Scholar] [CrossRef][Green Version]
- Thomson, E.; Rappsilber, J.; Tollervey, D. Nop9 is an RNA binding protein present in pre-40S ribosomes and required for 18S rRNA synthesis in yeast. RNA 2007, 13, 2165–2174. [Google Scholar] [CrossRef]
- Gialluisi, A.; Newbury, D.F.; Wilcutt, E.G.; Olson, R.K.; DeFries, J.C.; Brandler, W.M.; Pennington, B.F.; Smith, S.D.; Scerri, T.S.; Simpson, N.H.; et al. Genome-wide screening for DNA variants associated with reading and language traits. Genes Brain Behav. 2014, 13, 686–701. [Google Scholar] [CrossRef]
- Jangi, M.; Boutz, P.L.; Paul, P.; Sharp, P.A. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014, 28, 637–651. [Google Scholar] [CrossRef]
- Frankiw, L.; Majumdar, D.; Burns, C.; Vlach, L.; Moradian, A.; Sweredoski, M.J.; Baltimore, D. BUD13 Promotes a Type I Interferon Response by Countering Intron Retention in Irf7. Mol. Cell 2019, 73, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.P.; Moreno-Mateos, M.A.; Gohr, A.; Miao, L.; Chan, S.H.; Irimia, M.; Giraldez, A.J. RES complex is associated with intron definition and required for zebrafish early embryogenesis. PLoS Genet. 2018, 14, e1007473. [Google Scholar] [CrossRef]
- Servetti, M.; Pisciotta, L.; Tassano, E.; Cerminara, M.; Nobili, L.; Boeri, S.; Rosti, G.; Lerone, M.; Divizia, M.T.; Ronchetto, P.; et al. Neurodevelopmental Disorders in Patients With Complex Phenotypes and Potential Complex Genetic Basis Involving Non-Coding Genes, and Double CNVs. Front. Genet. 2021, 12, 732002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; You, Y.; Wu, Y.; Zhang, Y.; Wang, M.; Song, Y.; Liu, X.; Kou, C. Association of BUD13 polymorphisms with metabolic syndrome in Chinese population: A case-control study. Lipids Health Dis. 2017, 16, 127. [Google Scholar] [CrossRef][Green Version]
- Lin, E.; Kuo, P.-H.; Liu, Y.-L.; Yang, A.C.; Kao, C.-F.; Tsai, S.-J. Association and interaction of APOA5, BUD13, CETP, LIPA and health-related behavior with metabolic syndrome in a Taiwanese population. Sci. Rep. 2016, 6, 36830. [Google Scholar] [CrossRef]
- Monteuuis, G.; Wong, J.J.L.; Bailey, C.G.; Schmitz, U.; Rasko, J.E.J. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef]
- Li, H.D.; Funk, C.C.; McFarland, K.; Dammer, E.B.; Allen, M.; Carrasquillo, M.M.; Levites, Y.; Chakrabarty, P.; Burgess, J.D.; Wang, X.; et al. Integrative functional genomic analysis of intron retention in human and mouse brain with Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 984–1004. [Google Scholar] [CrossRef]
- Fu, L.; Shi, Z.; Luo, G.; Tu, W.; Wang, X.; Fang, Z.; Li, X. Multiple microRNAs regulate human FOXP2 gene expression by targeting sequences in its 3′ untranslated region. Mol. Brain 2014, 7, 71. [Google Scholar] [CrossRef]
- Weil, D.; Piton, A.; Lessel, D.; Standart, N. Mutations in genes encoding regulators of mRNA decapping and translation initiation: Links to intellectual disability. Biochem. Soc. Trans. 2020, 48, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.L. Toward epigenetic and gene regulation models of specific language impairment: Looking for links among growth, genes, and impairments. J. Neurodev. Disord. 2012, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.-L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Yap, K.; Lim, Z.Q.; Khandelia, P.; Friedman, B.; Makeyev, E.V. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev. 2012, 26, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef]
- Thompson, M.; Bixby, R.; Dalton, R.; Vandenburg, A.; Calarco, J.A.; Norris, A.D. Splicing in a single neuron is coordinately controlled by RNA binding proteins and transcription factors. eLife 2019, 8, e46726. [Google Scholar] [CrossRef]


| Gene | Genomic Position (hg19) | c.DNA Variant | AA Change | rsID | IDs of SLI Probands with Variant n = 175 | MAF in gnomAD | In Silico Prediction Scores | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glob | Euro | SIFT | Poly Phen-2 | Mutation assessor | PROVEAN | Mutation Taster | ||||||
| KIAA0319 | Chr6: | c.2164G > A | p.Arg722Trp | rs113411083 | NA | 0.00275 | 0.0047 | 0.068 | 0.998 | 1.495 | −3.28 | 101 |
| 24566953 | (T) | prob D | (low) | D | P | |||||||
| FLNC | Chr7: | c.6808G > A | p.Glu2270Lys | rs202223616 | NA | 0.00073 | 0.00168 | 1 | 0.371 | 1.23 | −2.44 | 56 |
| 128494547 | (T) | B | (low) | N | DC | |||||||
| NOP9 | Chr14: | c.62G > C | p.Arg21Pro | rs183868211 | 346, 353, 355, 411, 472 | 0.00936 | 0.02304 | 0.147 | 0.01 | 2.39 | −0.94 | 103 |
| 24769222 | (T) | B | (med) | N | P | |||||||
| Gene | Genomic Position (hg19) | c.DNA Variant | AA Change | rsID | IDs of SLI Probands with Variant n = 175 | MAF in gnomAD | In Silico Prediction Scores | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glob | Euro | SIFT | Poly Phen-2 | Mutation Assessor | PROVEAN | Mutation Taster | ||||||
| BUD13 | Chr11: | c.689G > A | p.Arg230Glu | rs139478949 | NA | 0.00002 | 0.00005 | 0.013 | 0.934 | 3.405 | −2.51 | 43 |
| 116633616 | (D) | poss D | (med) | D | DC | |||||||
| APLP2 | Chr11: | c.2041G > A | p.Val681Met | rs370970986 | NA | 0.00002 | 0.00002 | 0.39 | 0.94 | 1.1 | −0.01 | 21 |
| 130011820 | (D) | poss D | (low) | N | P | |||||||
| NDRG2 | Chr14: | c.143G > A | p.Gly48Asp | rs11552412 | NA | NA | NA | 0 | 1.00 | 3.445 | −6.06 | 94 |
| 21490631 | (D) | prob D | (med) | D | DC | |||||||
| Genomic Position (hg19) Chr11 | c.DNA | AA Change | rsID | IDs of SLI Probands with Variant n = 175 | MAF in gnomAD | In Silico Prediction Scores | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Glob | Euro | SIFT | Poly Phen-2 | Mutation Assessor | PROVEAN | Mutation Taster | |||||
| 129991652 | c.660T > G | p.Asp220Glu | rs1063201 | 434 | 0.00006 | 0.00002 | 0.736 | 0 | 0.255 | −0.59 | 45 |
| (T) | B | (neutral) | N | P | |||||||
| 129992279 | c.793G > A | p.Glu265Lys | NA | 463 | NA | NA | 0.022 | 0 | 0.55 | −1.54 | 56 |
| (D) | B | (neutral) | N | DC | |||||||
| 130013358 | c.*15G > A | 3′UTR | rs201861910 | 447 | 0.001221 | 0.001972 | NA | NA | NA | NA | NA |
| Genomic Position (hg19) Chr11 | c.DNA | AA Change | rsID | IDs of SLI Probands with Variant n = 175 | MAF in gnomAD | In Silico Prediction Scores | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Glob | Euro | SIFT | Poly Phen-2 | Mutation Assessor | PROVEAN | Mutation Taster | |||||
| 116643617 | c.64C > A | p.Ala22Ser | rs35585096 | 337, 455, 483, 405 | 0.023 | 0.000 | 0.112 | 0.578 | 2.44 | −0.81 | 99 |
| (D) | poss D | (med) | N | P | |||||||
| 116633875 | c.430G > A | p.Arg144Cys | rs116087150 | 49324 | 0.000 | 0.000 | 0.045 | 1 | 2.81 | −4.15 | 180 |
| (D) | prob D | (med) | D | DC | |||||||
| 116633787 | c.518T > C | p.Asp173Gly | rs1467808735 | 360 | 0.000 | 0.000 | 0.013 | 1 | 3.27 | −4.47 | 94 |
| (D) | prob D | (med) | D | DC | |||||||
| 116633724 | c.581C > T | p.Arg194His | rs144776650 | 384, 422, 484 | 0.003 | 0.005 | 0.06 | 0.091 | 1.725 | −3.73 | 29 |
| (T) | B | (low) | D | P | |||||||
| 116633580 | c.725C > A | p.Arg242Ile | rs11216131 | 500 | 0.001 | 0.001 | 0.002 | 0.999 | 3.58 | −4.43 | 97 |
| (D) | prob D | (high) | D | DC | |||||||
| 116633425 | c.880C > G | p.Ala294Pro | rs1427011653 | 201 | NA | NA | 0.231 | 0.002 | 2.395 | −0.75 | 27 |
| (T) | B | (med) | N | P | |||||||
| 116633353 | c.952A > T | p.Tyr318Asn | rs145410701 | 438 | 0.001 | 0.000 | 0.33 | 0.138 | 2.045 | −1.26 | 142 |
| (T) | B | (med) | N | P | |||||||
| 116631482 | c.1223G > A | p.Pro408Leu | rs61730763 | 427 | 0.003 | 0.000 | 0.023 | 0.275 | 2.63 | −7.04 | 98 |
| (D) | B | (med) | D | DC | |||||||
| 116619178 | c.*20G > A | 3′UTR | rs145906707 | 431, 447 | 0.003 | 0.003 | NA | NA | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andres, E.M.; Earnest, K.K.; Zhong, C.; Rice, M.L.; Raza, M.H. Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex. Brain Sci. 2022, 12, 47. https://doi.org/10.3390/brainsci12010047
Andres EM, Earnest KK, Zhong C, Rice ML, Raza MH. Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex. Brain Sciences. 2022; 12(1):47. https://doi.org/10.3390/brainsci12010047
Chicago/Turabian StyleAndres, Erin M., Kathleen Kelsey Earnest, Cuncong Zhong, Mabel L. Rice, and Muhammad Hashim Raza. 2022. "Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex" Brain Sciences 12, no. 1: 47. https://doi.org/10.3390/brainsci12010047
APA StyleAndres, E. M., Earnest, K. K., Zhong, C., Rice, M. L., & Raza, M. H. (2022). Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex. Brain Sciences, 12(1), 47. https://doi.org/10.3390/brainsci12010047