
Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature

 ,
, 
Abstract
1. Introduction
2. Methods
3. Results
3.1. Neurocognitive Correlates and Illness Neuroprogression
3.2. Neuroimaging Evidence and Neuroprogression in BD: An Exciting Journey through the History of This Link
3.3. BD and Neurodegeneration: The Role of Lithium
4. Neural Circuits of BD as Revealed by Neuroimaging Techniques: A Comprehensive Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Carvalho, A.F.; Firth, J.; Vieta, E. Bipolar Disorder. N. Engl. J. Med. 2020, 383, 58–66. [Google Scholar] [CrossRef]
- Parker, G.; McCraw, S.; Hadzi-Pavlovic, D.; Fletcher, K. Costs of the principal mood disorders: A study of comparative direct and indirect costs incurred by those with bipolar I, bipolar II and unipolar disorders. J. Affect. Disord. 2013, 149, 46–55. [Google Scholar] [CrossRef]
- Parker, G.; Roy, K.; Wilhelm, K.; Mitchell, P.; Hadzi-Pavlovic, D. The nature of bipolar depression: Implications for the definition of melancholia. J. Affect. Disord. 2000, 59, 217–224. [Google Scholar] [CrossRef]
- Ruiz, N.A.L.; Del Ángel, D.S.; Olguín, H.J.; Silva, M.L. Neuroprogression: The hidden mechanism of depression. Neuropsychiatr. Dis. Treat. 2018, 14, 2837. [Google Scholar] [CrossRef] [PubMed]
- Kapczinski, N.S.; Mwangi, B.; Cassidy, R.M.; Librenza-Garcia, D.; Bermudez, M.B.; Kauer-Sant’anna, M.; Kapczinski, F.; Passos, I.C. Neuroprogression and illness trajectories in bipolar disorder. Expert Rev. Neurother. 2017, 17, 277–285. [Google Scholar] [CrossRef]
- Shioya, A.; Saito, Y.; Arima, K.; Kakuta, Y.; Yuzuriha, T.; Tanaka, N.; Murayama, S.; Tamaoka, A. Neurodegenerative changes in patients with clinical history of bipolar disorders. Neuropathology 2015, 35, 245–253. [Google Scholar] [CrossRef]
- Tohen, M.; Hennen, J.; Zarate, C.M., Jr.; Baldessarini, R.J.; Strakowski, S.M.; Stoll, A.L.; Faedda, G.L.; Suppes, T.; Gebre-Medhin, P.; Cohen, B.M. Two-year syndromal and functional recovery in 219 cases of first-episode major affective disorder with psychotic features. Am. J. Psychiatry 2000, 157, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Kapczinski, F.; Andreazza, A.C.; Dean, O.M.; Giorlando, F.; Maes, M.; Yücel, M.; Gama, C.S.; Dodd, S.; Dean, B.; et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011, 35, 804–817. [Google Scholar] [CrossRef]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Bearden, C.E.; Hoffman, K.M.; Cannon, T.D. The neuropsychology and neuroanatomy of bipolar affective disorder: A critical review. Bipolar Disord. 2001, 3, 106–150. [Google Scholar] [CrossRef]
- Vavakova, M.; Durackova, Z.; Trebaticka, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxidative Med. Cell. Longev. 2015, 2015, 898393. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.B.; Frey, B.N.; Andreazza, A.C.; Goi, J.D.; Rosa, A.R.; Gonçalves, C.A.; Santin, A.; Kapczinski, F. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci. Lett. 2006, 398, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, B.S.; Gama, C.S.; Cereser, K.M.; Yatham, L.N.; Fries, G.R.; Colpo, G.; de Lucena, D.; Kunz, M.; Gomes, F.A.; Kapczinski, F. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: A systematic review and meta-regression analysis. J. Psychiatr. Res. 2011, 45, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Gigante, A.D.; Young, L.T.; Yatham, L.N.; Andreazza, A.C.; Nery, F.G.; Grinberg, L.T.; Heinsen, H.; Lafer, B. Morphometric post-mortem studies in bipolar disorder: Possible association with oxidative stress and apoptosis. Int. J. Neuropsychopharmacol. 2011, 14, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Kapczinski, F.; Frey, B.N.; Kauer-Sant’Anna, M.; Grassi-Oliveira, R. Brain-derived neurotrophic factor and neuroplasticity in bipolar disorder. Expert. Rev. Neurother. 2008, 8, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Kapczinski, F.; Dal-Pizzol, F.; Teixeira, A.L.; Magalhaes, P.V.; Kauer-Sant’Anna, M.; Klamt, F.; Pasquali, M.A.; Quevedo, J.; Gama, C.S.; Post, R. A systemic toxicity index developed to assess peripheral changes in mood episodes. Mol. Psychiatry 2010, 15, 784–786. [Google Scholar] [CrossRef]
- Nery, F.G.; Gigante, A.D.; Amaral, J.A.; Fernandes, F.B.; Berutti, M.; Almeida, K.M.; Stertz, L.; Bristot, G.; Kapczinski, F.; Lafer, B. Serum BDNF levels in unaffected first-degree relatives of patients with bipolar disorder. Braz. J. Psychiatry 2016, 38, 197–200. [Google Scholar] [CrossRef]
- Passos, I.C.; Mwangi, B.; Vieta, E.; Berk, M.; Kapczinski, F. Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatr. Scand. 2016, 134, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Tidemalm, D.; Haglund, A.; Karanti, A.; Landén, M.; Runeson, B. Attempted suicide in bipolar disorder: Risk factors in a cohort of 6086 patients. PLoS ONE 2014, 9, e94097. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.; Horrocks, J.; Doucette, S.; Keown-Stoneman, C.; McCloskey, S.; Grof, P. The developmental trajectory of bipolar disorder. Br. J. Psychiatry 2014, 204, 122–128. [Google Scholar] [CrossRef]
- Muneer, A. The Neurobiology of Bipolar Disorder: An Integrated Approach. Chonnam Med. J. 2016, 52, 18–37. [Google Scholar] [CrossRef]
- Latalova, K.; Prasko, J.; Diveky, T.; Velartova, H. Cognitive impairment in bipolar disorder. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2011, 155, 19–26. [Google Scholar] [CrossRef]
- Martínez-Arán, A.; Vieta, E.; Colom, F.; Torrent, C.; Sánchez-Moreno, J.; Reinares, M.; Benabarre, A.; Goikolea, J.M.; Brugué, E.; Daban, C. Cognitive impairment in euthymic bipolar patients: Implications for clinical and functional outcome. Bipolar Disord. 2004, 6, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Fournier, J.C.; Chase, H.W.; Almeida, J.; Phillips, M.L. Within- and Between-Session Changes in Neural Activity During Emotion Processing in Unipolar and Bipolar Depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 518–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clark, L.; Iversen, S.D.; Goodwin, G.M. Sustained attention deficit in bipolar disorder. Br. J. Psychiatry 2002, 180, 313–319. [Google Scholar] [CrossRef]
- Deckersbach, T.; McMurrich, S.; Ogutha, J.; Savage, C.R.; Sachs, G.; Rauch, S.L. Characteristics of non-verbal memory impairment in bipolar disorder: The role of encoding strategies. Psychol. Med. 2004, 34, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.J.; Ferrier, I.N. Evolution of cognitive impairment in bipolar disorder: A systematic review of cross-sectional evidence. Bipolar Disord. 2006, 8, 103–116. [Google Scholar] [CrossRef]
- Torres, I.J.; DeFreitas, V.G.; DeFreitas, C.M.; Kauer-Sant’Anna, M.; Bond, D.J.; Honer, W.G.; Lam, R.W.; Yatham, L.N. Neurocognitive functioning in patients with bipolar I disorder recently recovered from a first manic episode. J. Clin. Psychiatry 2010, 71, 1234–1242. [Google Scholar] [CrossRef]
- Gama, C.S.; Kunz, M.; Magalhães, P.V.; Kapczinski, F. Staging and neuroprogression in bipolar disorder: A systematic review of the literature. Braz. J. Psychiatry 2013, 35, 70–74. [Google Scholar] [CrossRef]
- Kapczinski, F.; Magalhães, P.V.; Balanzá-Martinez, V.; Dias, V.V.; Frangou, S.; Gama, C.S.; Gonzalez-Pinto, A.; Grande, I.; Ha, K.; Kauer-Sant’Anna, M. Staging systems in bipolar disorder: An International Society for Bipolar Disorders Task Force Report. Acta Psychiatr. Scand. 2014, 130, 354–363. [Google Scholar] [CrossRef]
- Post, R.M.; Fleming, J.; Kapczinski, F. Neurobiological correlates of illness progression in the recurrent affective disorders. J. Psychiatr. Res. 2012, 46, 561–573. [Google Scholar] [CrossRef]
- Rodrigues, A.A.; Rosa, A.R.; Kunz, M.; Bruna, A.; Kapczinski, F. Bipolar disorder: Staging and neuroprogression. Psychiatr. Pol. 2014, 48, 231–243. [Google Scholar]
- Vieta, E.; Popovic, D.; Rosa, A.R.; Solé, B.; Grande, I.; Frey, B.N.; Martinez-Aran, A.; Sanchez-Moreno, J.; Balanzá-Martínez, V.; Tabarés-Seisdedos, R.; et al. The clinical implications of cognitive impairment and allostatic load in bipolar disorder. Eur. Psychiatry 2013, 28, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Post, R.; Ratheesh, A.; Gliddon, E.; Singh, A.; Vieta, E.; Carvalho, A.F.; Ashton, M.M.; Berk, L.; Cotton, S.M.; et al. Staging in bipolar disorder: From theoretical framework to clinical utility. World Psychiatry 2017, 16, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Fries, G.R.; Pfaffenseller, B.; Stertz, L.; Paz, A.V.C.; Dargél, A.A.; Kunz, M.; Kapczinski, F. Staging and neuroprogression in bipolar disorder. Curr. Psychiatry Rep. 2012, 14, 667–675. [Google Scholar] [CrossRef]
- Berk, M. Neuroprogression: Pathways to progressive brain changes in bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, P.V.; Dodd, S.; Nierenberg, A.A.; Berk, M. Cumulative morbidity and prognostic staging of illness in the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Aust. N. Z. J. Psychiatry 2012, 46, 1058–1067. [Google Scholar] [CrossRef]
- Kozicky, J.M.; Torres, I.J.; Silveira, L.E.; Bond, D.J.; Lam, R.W.; Yatham, L.N. Cognitive change in the year after a first manic episode: Association between clinical outcome and cognitive performance early in the course of bipolar I disorder. J. Clin. Psychiatry. 2014, 75, e587–e593. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S.; Teixeira, A.L.; Cao, F.; Gildengers, A.; Soares, J.C.; Butters, M.A.; Reynolds, C.F., 3rd. History of Bipolar Disorder and the Risk of Dementia: A Systematic Review and Meta-Analysis. Am. J. Geriatr. Psychiatry 2017, 25, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Velosa, J.; Delgado, A.; Finger, E.; Berk, M.; Kapczinski, F.; de Azevedo Cardoso, T. Risk of dementia in bipolar disorder and the interplay of lithium: A systematic review and meta-analyses. Acta Psychiatr. Scand. 2020, 141, 510–521. [Google Scholar] [CrossRef]
- El-Badri, S.M.; Ashton, C.H.; Moore, P.B.; Marsh, V.R.; Ferrier, I.N. Electrophysiological and cognitive function in young euthymic patients with bipolar affective disorder. Bipolar Disord. 2001, 3, 79–87. [Google Scholar] [CrossRef]
- Kieseppä, T.; Tuulio-Henriksson, A.; Haukka, J.; Van Erp, T.; Glahn, D.; Cannon, T.D.; Partonen, T.; Kaprio, J.; Lönnqvist, J. Memory and verbal learning functions in twins with bipolar I disorder, and the role of information-processing speed. Psychol. Med. 2005, 35, 205. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.J.; Strejilevich, S.A.; Marengo, E.; Igoa, A.; Fassi, G.; Teitelbaum, J.; Caravotta, P. Relationship between neurocognitive functioning and episode recurrences in bipolar disorder. J. Affect. Disord. 2013, 147, 345–351. [Google Scholar] [CrossRef]
- Sparding, T.; Silander, K.; Pålsson, E.; Östlind, J.; Ekman, C.J.; Sellgren, C.M.; Joas, E.; Hansen, S.; Landén, M. Classification of cognitive performance in bipolar disorder. Cogn. Neuropsychiatry 2017, 22, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Gildengers, A.G.; Chisholm, D.; Butters, M.A.; Anderson, S.J.; Begley, A.; Holm, M.; Rogers, J.C.; Reynolds, C.F., 3rd; Mulsant, B.H. Two-year course of cognitive function and instrumental activities of daily living in older adults with bipolar disorder: Evidence for neuroprogression? Psychol. Med. 2013, 43, 801–811. [Google Scholar] [CrossRef]
- Samamé, C.; Martino, D.J.; Strejilevich, S.A. Longitudinal course of cognitive deficits in bipolar disorder: A meta-analytic study. J. Affect. Disord. 2014, 164, 130–138. [Google Scholar] [CrossRef]
- Cullen, B.; Ward, J.; Graham, N.A.; Deary, I.J.; Pell, J.P.; Smith, D.J.; Evans, J.J. Prevalence and correlates of cognitive impairment in euthymic adults with bipolar disorder: A systematic review. J. Affect. Disord. 2016, 205, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Szmulewicz, A.G.; Valerio, M.P.; Smith, J.M.; Samamé, C.; Martino, D.J.; Strejilevich, S.A. Neuropsychological profiles of major depressive disorder and bipolar disorder during euthymia. A systematic literature review of comparative studies. Psychiatry Res. 2017, 248, 127–133. [Google Scholar] [CrossRef]
- Vlad, M.; Raucher-Chéné, D.; Henry, A.; Kaladjian, A. Functional outcome and social cognition in bipolar disorder: Is there a connection? Eur. Psychiatry 2018, 52, 116–125. [Google Scholar] [CrossRef]
- Vierck, E.; Porter, R.J.; Joyce, P.R. Facial recognition deficits as a potential endophenotype in bipolar disorder. Psychiatry Res. 2015, 230, 102–107. [Google Scholar] [CrossRef]
- Benito, A.; Lahera, G.; Herrera, S.; Muncharaz, R.; Benito, G.; Fernández-Liria, A.; Montes, J.M. Deficits in recognition, identification, and discrimination of facial emotions in patients with bipolar disorder. Braz. J. Psychiatry 2013, 35, 435–438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lahera, G.; Herrera, S.; Reinares, M.; Benito, A.; Rullas, M.; González-Cases, J.; Vieta, E. Hostile attributions in bipolar disorder and schizophrenia contribute to poor social functioning. Acta Psychiatr. Scand. 2015, 131, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Belizario, G.O.; Gigante, A.D.; de Almeida Rocca, C.C.; Lafer, B. Cognitive impairments and predominant polarity in bipolar disorder: A cross-sectional study. Int. J. Bipolar Disord. 2017, 5, 15. [Google Scholar] [CrossRef]
- Librenza-Garcia, D.; Kotzian, B.J.; Yang, J.; Mwangi, B.; Cao, B.; Pereira Lima, L.N.; Bermudez, M.B.; Boeira, M.V.; Kapczinski, F.; Passos, I.C. The impact of machine learning techniques in the study of bipolar disorder: A systematic review. Neurosci. Biobehav. Rev. 2017, 80, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.J.; Mwangi, B.; Bauer, I.E.; Passos, I.C.; Sanches, M.; Zunta-Soares, G.B.; Meyer, T.D.; Hasan, K.M.; Soares, J.C. Identification and individualized prediction of clinical phenotypes in bipolar disorders using neurocognitive data, neuroimaging scans and machine learning. Neuroimage 2017, 145, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Bora, E.; Fornito, A.; Yucel, M.; Pantelis, C. Voxelwise meta-analysis of gray matter abnormalities in bipolar disorder. Biol. Psychiatry 2010, 67, 1097–1105. [Google Scholar] [CrossRef]
- Lin, A.; Reniers, R.L.; Wood, S.J. Clinical staging in severe mental disorder: Evidence from neurocognition and neuroimaging. Br. J. Psychiatry 2013, 54, s11–s17. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.O.; Mann, L.S.; Weinberger, D.R.; van Kammen, D.P.; Post, R.M. Computed tomographic scans in patients with schizophrenia, schizoaffective, and bipolar affective disorder. Arch. Gen. Psychiatry 1983, 40, 735–739. [Google Scholar] [CrossRef]
- Schneider, M.R.; DelBello, M.P.; McNamara, R.K.; Strakowski, S.M.; Adler, C.M. Neuroprogression in bipolar disorder. Bipolar Disord. 2012, 14, 356–374. [Google Scholar] [CrossRef]
- Hallahan, B.; Newell, J.; Soares, J.C.; Brambilla, P.; Strakowski, S.M.; Fleck, D.E.; Kieseppä, T.; Altshuler, L.L.; Fornito, A.; Malhi, G.S. Structural magnetic resonance imaging in bipolar disorder: An international collaborative mega-analysis of individual adult patient data. Biol. Psychiatry. 2011, 69, 326–335. [Google Scholar] [CrossRef]
- Lim, C.S.; Baldessarini, R.J.; Vieta, E.; Yucel, M.; Bora, E.; Sim, K. Longitudinal neuroimaging and neuropsychological changes in bipolar disorder patients: Review of the evidence. Neurosci. Biobehav. Rev. 2013, 37, 418–435. [Google Scholar] [CrossRef]
- Arslan, S.; Ktena, S.I.; Makropoulos, A.; Robinson, E.C.; Rueckert, D.; Parisot, S. Human brain mapping: A systematic comparison of parcellation methods for the human cerebral cortex. Neuroimage 2018, 170, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, J.H.; Wang, F.; Spencer, L.; Edmiston, E.; Lacadie, C.M.; Martin, A.; Constable, R.T.; Duncan, J.S.; Staib, L.H.; Papademetris, X.; et al. Preliminary evidence for progressive prefrontal abnormalities in adolescents and young adults with bipolar disorder. J. Int. Neuropsychol. Soc. 2009, 15, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Arango, C.; Rapado-Castro, M.; Reig, S.; Castro-Fornieles, J.; González-Pinto, A.; Otero, S.; Baeza, I.; Moreno, C.; Graell, M.; Janssen, J.; et al. Progressive brain changes in children and adolescents with first-episode psychosis. Arch. Gen. Psychiatry 2012, 69, 16–26. [Google Scholar] [CrossRef]
- Gogtay, N.; Ordonez, A.; Herman, D.H.; Hayashi, K.M.; Greenstein, D.; Vaituzis, C.; Lenane, M.; Clasen, L.; Sharp, W.; Giedd, J.N. Dynamic mapping of cortical development before and after the onset of pediatric bipolar illness. J. Child Psychol. Psychiatry 2007, 48, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Lisy, M.E.; Jarvis, K.B.; DelBello, M.P.; Mills, N.P.; Weber, W.A.; Fleck, D.; Strakowski, S.M.; Adler, C.M. Progressive neurostructural changes in adolescent and adult patients with bipolar disorder. Bipolar Disord. 2011, 13, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Harenski, K.; Nicoletti, M.; Sassi, R.B.; Mallinger, A.G.; Frank, E.; Kupfer, D.J.; Keshavan, M.S.; Soares, J.C. MRI investigation of temporal lobe structures in bipolar patients. J. Psychiatr. Res. 2003, 37, 287–295. [Google Scholar] [CrossRef]
- Moorhead, T.W.; McKirdy, J.; Sussmann, J.E.; Hall, J.; Lawrie, S.M.; Johnstone, E.C.; McIntosh, A.M. Progressive gray matter loss in patients with bipolar disorder. Biol. Psychiatry 2007, 62, 894–900. [Google Scholar] [CrossRef]
- Dusi, N.; De Carlo, V.; Delvecchio, G.; Bellani, M.; Soares, J.C.; Brambilla, P. MRI features of clinical outcome in bipolar disorder: A selected review. J. Affect. Disord. 2019, 243, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Doris, A.; Belton, E.; Ebmeier, K.P.; Glabus, M.F.; Marshall, I. Reduction of cingulate gray matter density in poor outcome bipolar illness. Psychiatry Res. 2004, 130, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Nanda, P.; Tandon, N.; Mathew, I.T.; Padmanabhan, J.L.; Clementz, B.A.; Pearlson, G.D.; Sweeney, J.A.; Tamminga, C.A.; Keshavan, M.S. Impulsivity across the psychosis spectrum: Correlates of cortical volume, suicidal history, and social and global function. Schizophr. Res. 2016, 170, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Knöchel, C.; Stäblein, M.; Prvulovic, D.; Ghinea, D.; Wenzler, S.; Pantel, J.; Alves, G.; Linden, D.E.; Harrison, O.; Carvalho, A.; et al. Shared and distinct gray matter abnormalities in schizophrenia, schizophrenia relatives and bipolar disorder in association with cognitive impairment. Schizophr. Res. 2016, 171, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Lista, S.; Mango, D.; Nisticò, R.; Perry, G.; Avila, J.; Hernandez, F.; Geerts, H.; Vergallo, A.; Alzheimer Precision Medicine Initiative (APMI). Lithium as a Treatment for Alzheimer’s Disease: The Systems Pharmacology Perspective. J. Alzheimers Dis. 2019, 69, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zeng, W.Z.; Yuan, P.X.; Huang, L.D.; Jiang, Y.M.; Zhao, Z.H.; Manji, H.K. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 1999, 72, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Moore, G.J.; Chen, G. Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: A role for neurotrophic and neuroprotective effects in manic depressive illness. J. Clin. Psychiatry 2000, 61, 82–96. [Google Scholar] [PubMed]
- Muneer, A. Wnt and GSK3 Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications. Clin. Psychopharmacol. Neurosci. 2017, 15, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G.S.; Tanious, M.; Das, P.; Coulston, C.M.; Berk, M. Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs 2013, 27, 135–153. [Google Scholar] [CrossRef]
- Hashimoto, R.; Hough, C.; Nakazawa, T.; Yamamoto, T.; Chuang, D.M. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: Involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 2002, 80, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Haroutunian, V.; Katsel, P.; Roussos, P.; Davis, K.L.; Altshuler, L.L.; Bartzokis, G. Myelination, oligodendrocytes, and serious mental illness. Glia 2014, 62, 1856–1877. [Google Scholar] [CrossRef]
- Emsell, L.; Langan, C.; Van Hecke, W.; Barker, G.J.; Leemans, A.; Sunaert, S.; McCarthy, P.; Nolan, R.; Cannon, D.M.; McDonald, C. White matter differences in euthymic bipolar I disorder: A combined magnetic resonance imaging and diffusion tensor imaging voxel-based study. Bipolar Disord. 2013, 15, 365–376. [Google Scholar] [CrossRef]
- Mosebach, J.; Keilhoff, G.; Gos, T.; Schiltz, K.; Schoeneck, L.; Dobrowolny, H.; Mawrin, C.; Muller, S.; Schroeter, M.L.; Bernstein, H.G.; et al. Increased nuclear Olig1-expression in the pregenual anterior cingulate white matter of patients with major depression: A regenerative attempt to compensate oligodendrocyte loss? J. Psychiatr. Res. 2013, 47, 1069–1079. [Google Scholar] [CrossRef]
- Sassi, R.B.; Brambilla, P.; Hatch, J.P.; Nicoletti, M.A.; Mallinger, A.G.; Frank, E.; Kupfer, D.J.; Keshavan, M.S.; Soares, J.C. Reduced left anterior cingulate volumes in untreated bipolar patients. Biol. Psychiatry 2004, 56, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Sassi, R.B.; Nicoletti, M.; Brambilla, P.; Mallinger, A.G.; Frank, E.; Kupfer, D.J.; Keshavan, M.S.; Soares, J.C. Increased gray matter volume in lithium-treated bipolar disorder patients. Neurosci. Lett. 2002, 329, 243–245. [Google Scholar] [CrossRef]
- Moore, G.J.; Bebchuk, J.M.; Hasanat, K.; Chen, G.; Seraji-Bozorgzad, N.; Wilds, I.B.; Faulk, M.W.; Koch, S.; Glitz, D.A.; Jolkovsky, L.; et al. Lithium increases N-acetyl-aspartate in the human brain: In vivo evidence in support of bcl-2′s neurotrophic effects? Biol. Psychiatry 2000, 48, 1–8. [Google Scholar] [CrossRef]
- Yucel, K.; McKinnon, M.C.; Taylor, V.H.; Macdonald, K.; Alda, M.; Young, L.T.; MacQueen, G.M. Bilateral hippocampal volume increases after long-term lithium treatment in patients with bipolar disorder: A longitudinal MRI study. Psychopharmacology (Berl.) 2007, 195, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, M. Lithium side effects and toxicity: Prevalence and management strategies. Int. J. Bipolar. Disord. 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Burdick, K.E.; Millett, C.E.; Russo, M.; Alda, M.; Alliey-Rodriguez, N.; Anand, A.; Balaraman, Y.; Berrettini, W.; Bertram, H.; Calabrese, J.R.; et al. The association between lithium use and neurocognitive performance in patients with bipolar disorder. Neuropsychopharmacology 2020, 45, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Sabater, A.; García-Blanco, A.C.; Verdet, H.M.; Sierra, P.; Ribes, J.; Villar, I.; Lara, M.J.; Arnal, P.; Rojo, L.; Livianos, L. Comparative neurocognitive effects of lithium and anticonvulsants in long-term stable bipolar patients. J. Affect. Disord. 2016, 190, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Dotson, V.M.; Beydoun, M.A.; Zonderman, A.B. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology 2010, 75, 27–34. [Google Scholar] [CrossRef]
- Saczynski, J.S.; Beiser, A.; Seshadri, S.; Auerbach, S.; Wolf, P.A. Depressive symptoms and risk of dementia: The Framingham heart study. Neurology 2010, 75, 35–41. [Google Scholar] [CrossRef]
- Strakowski, S.M.; Adler, C.M.; Almeida, J.; Altshuler, L.L.; Blumberg, H.P.; Chang, K.D.; DelBello, M.P.; Frangou, S.; McIntosh, A.; Phillips, M.L.; et al. The functional neuroanatomy of bipolar disorder: A consensus model. Bipolar Disord. 2012, 14, 313–325. [Google Scholar] [CrossRef]
- Hibar, D.P.; Westlye, L.T.; Doan, N.T.; Jahanshad, N.; Cheung, J.W.; Ching, C.R.K.; Versace, A.; Bilderbeck, A.C.; Uhlmann, A.; Mwangi, B. Cortical abnormalities in bipolar disorder: An MRI analysis of 6503 individuals from the ENIGMA bipolar disorder working group. Mol. Psychiatry 2017, 23, 932–942. [Google Scholar] [CrossRef]
- Blond, B.N.; Fredericks, C.A.; Blumberg, H.P. Functional neuroanatomy of bipolar disorder: Structure, function, and connectivity in an amygdala-anterior paralimbic neural system. Bipolar Disord. 2012, 14, 340–355. [Google Scholar] [CrossRef]
- Hibar, D.; Westlye, L.T.; Van Erp, T.; Rasmussen, J.; Leonardo, C.D.; Faskowitz, J.; Haukvik, U.K.; Hartberg, C.B.; Doan, N.T.; Agartz, I.; et al. Subcortical volumetric abnormalities in bipolar disorder. Mol. Psychiatry. 2016, 21, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Frangou, S. Brain structural and functional correlates of resilience to bipolar disorder. Front. Hum. Neurosci. 2012, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Morris, H.M.; Drevets, W.C. Neuroimaging studies of bipolar depression: Therapeutic implications. In Bipolar Depression: Molecular Neurobiology, Clinical Diagnosis, and Pharmacotherapy; Zarate, C.A., Manji, H.K., Eds.; Springer: Cham, Switzerland, 2016; pp. 137–181. [Google Scholar]
- El-Mallakh, R.S. Bipolar (manic depressive) disorders. In Psychiatry, 4th ed.; Tasman, A., Kay, J., Lieberman, J.A., First, M.B., Riba, M.B., Eds.; Wiley Blackwell: New York, NY, USA, 2015; pp. 868–872. [Google Scholar]
- Abé, C.; Ekman, C.-J.; Sellgren, C.; Petrovic, P.; Ingvar, M.; Landén, M. Manic episodes are related to changes in frontal cortex: A longitudinal neuroimaging study of bipolar disorder 1. Brain 2015, 138, 3440–3448. [Google Scholar] [CrossRef] [PubMed]
- Abé, C.; Rolstad, S.; Petrovic, P.; Ekman, C.-J.; Sparding, T.; Ingvar, M.; Landén, M. Bipolar disorder type I and II show distinct relationships between cortical thickness and executive function. Acta Psychiatr. Scand. 2018, 138, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Passos, I.C.; Mwangi, B.; Bauer, I.E.; Zunta-Soares, G.B.; Kapczinski, F.; Soares, J.C. Hippocampal volume and verbal memory performance in late-stage bipolar disorder. J. Psychiatr. Res. 2016, 73, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Haukvik, U.K.; Westlye, L.T.; Mørch-Johnsen, L.; Jørgensen, K.N.; Lange, E.H.; Dale, A.M.; Melle, I.; Andreassen, O.A.; Agartz, I. In Vivo Hippocampal subfield volumes in schizophrenia and bipolar disorder. Biol. Psychiatry 2015, 77, 581–588. [Google Scholar] [CrossRef]
- Mathew, I.; Gardin, T.M.; Tandon, N.; Eack, S.; Francis, A.N.; Seidman, L.J.; Clementz, B.; Pearlson, G.D.; Sweeney, J.A.; Tamminga, C.A.; et al. Medial temporal lobe structures and hippocampal subfields in psychotic disorders: Findings from the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. JAMA Psychiatry 2014, 71, 769–777. [Google Scholar] [CrossRef]
- Han, K.M.; Kim, A.; Kang, W.; Kang, Y.; Kang, J.; Won, E.; Tae, W.S.; Ham, B.J. Hippocampal subfield volumes in major depressive disorder and bipolar disorder. Eur. Psychiatry. 2019, 57, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.P.; Biggs, A.; Chen, G.; Pine, D.S.; Grillon, C. Contextual fear conditioning in humans: Cortical-hippocampal and amygdala contributions. J. Neurosci. 2008, 28, 6211–6219. [Google Scholar] [CrossRef]
- Phillips, M.L.; Drevets, W.C.; Rauch, S.L.; Lane, R. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biol. Psychiatry 2003, 54, 504–514. [Google Scholar] [CrossRef]
- MacDonald, A.W.; Cohen, J.D.; Stenger, V.A.; Carter, C.S. Dissociating the role of the dorsolateral prefrontal and anterior cingulate cortex in cognitive control. Science 2000, 288, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.D.; Bookheimer, S.Y.; Foland-Ross, L.C.; Moody, T.D.; Eisenberger, N.I.; Fischer, J.S.; Cohen, M.S.; Sugar, C.A.; Altshuler, L.L. Deficits in inferior frontal cortex activation in euthymic bipolar disorder patients during a response inhibition task. Bipolar Disord. 2012, 14, 442–450. [Google Scholar] [CrossRef]
- Gruber, S.A.; Rogowska, J.; Yurgelun-Todd, D.A. Decreased activation of the anterior cingulate in bipolar patients: An fMRI study. J. Affect. Disord. 2004, 82, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Hennings, J.M.; Owashi, T.; Binder, E.B.; Horstmann, S.; Menke, A.; Kloiber, S.; Wollweber, B.; Spieler, D.; Messer, T.; Lutz, R.; et al. Clinical characteristics and treatment outcome in a representative sample of depressed inpatients—Findings from the Munich antidepressant response signature (MARS) project. J. Psychiatr. Res. 2009, 43, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Librenza-Garcia, D.; Suh, J.S.; Watts, D.P.; Ballester, P.L.; Minuzzi, L.; Kapczinski, F.; Frey, B.N. Free Structural and functional brain correlates of neuroprogression in bipolar disorders. Curr. Top. Behav. Neurosci. 2020, in press. [Google Scholar]
- Besga, A.; Termenon, M.; Grana, M.; Echeveste, J.; Perez, J.M.; Gonzales Pinto, A. Discovering Alzheimer’s disease and bipolar disorder white matter effects building computer aided diagnostic systems on brain diffusion tensor imaging features. Neurosci. Lett. 2012, 520, 71–76. [Google Scholar] [CrossRef]
- Monkul, E.S.; Hatch, J.P.; Sassi, R.B.; Axelson, D.; Brambilla, P.; Nicoletti, M.A.; Keshavan, M.S.; Ryan, N.D.; Birmaher, B.; Soares, J.C. MRI study of the cerebellum in young bipolar patients. Progr. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 613–619. [Google Scholar] [CrossRef]
- Gokcinar, N.B.; Buturak, S.V.; Ozkal, F.; Ozcicek, G.; Yumusak, M.E.; Turgal, E. Optical coherence tomography neurodegenerative findings in patinets with bipolar disorders. Asia Pac. Psychiatry. 2020, 12, e12394. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.C.; Miller, B.L. Frontotemporal dementia. Semin. Neurol. 2013, 33, 336–341. [Google Scholar] [PubMed]
- Cea-Cañas, B.; Gomez-Pilar, J.; Núñez, P.; Rodríguez-Vázquez, E.; de Uribe, N.; Díez, Á.; Pérez-Escudero, A.; Molina, V. Connectivity strength of the EEG functional network in schizophrenia and bipolar disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 98, 109801. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O. Networks of the Brain; MIT Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Pompili, M.; Serafini, G.; Innamorati, M.; Venturini, P.; Fusar-Poli, P.; Sher, L.; Amore, M.; Girardi, P. Agomelatine, a novel intriguing antidepressant option enhancing neuroplasticity: A critical review. World J. Biol. Psychiatry 2013, 14, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Girardi, P.; Pompili, M.; Innamorati, M.; Mancini, M.; Serafini, G.; Mazzarini, L.; Del Casale, A.; Tatarelli, R.; Baldessarini, R.J. Duloxetine in acute major depression: Review of comparisons to placebo and standard antidepressants using dissimilar methods. Hum. Psychopharmacol. 2009, 24, 177–190. [Google Scholar] [CrossRef] [PubMed]


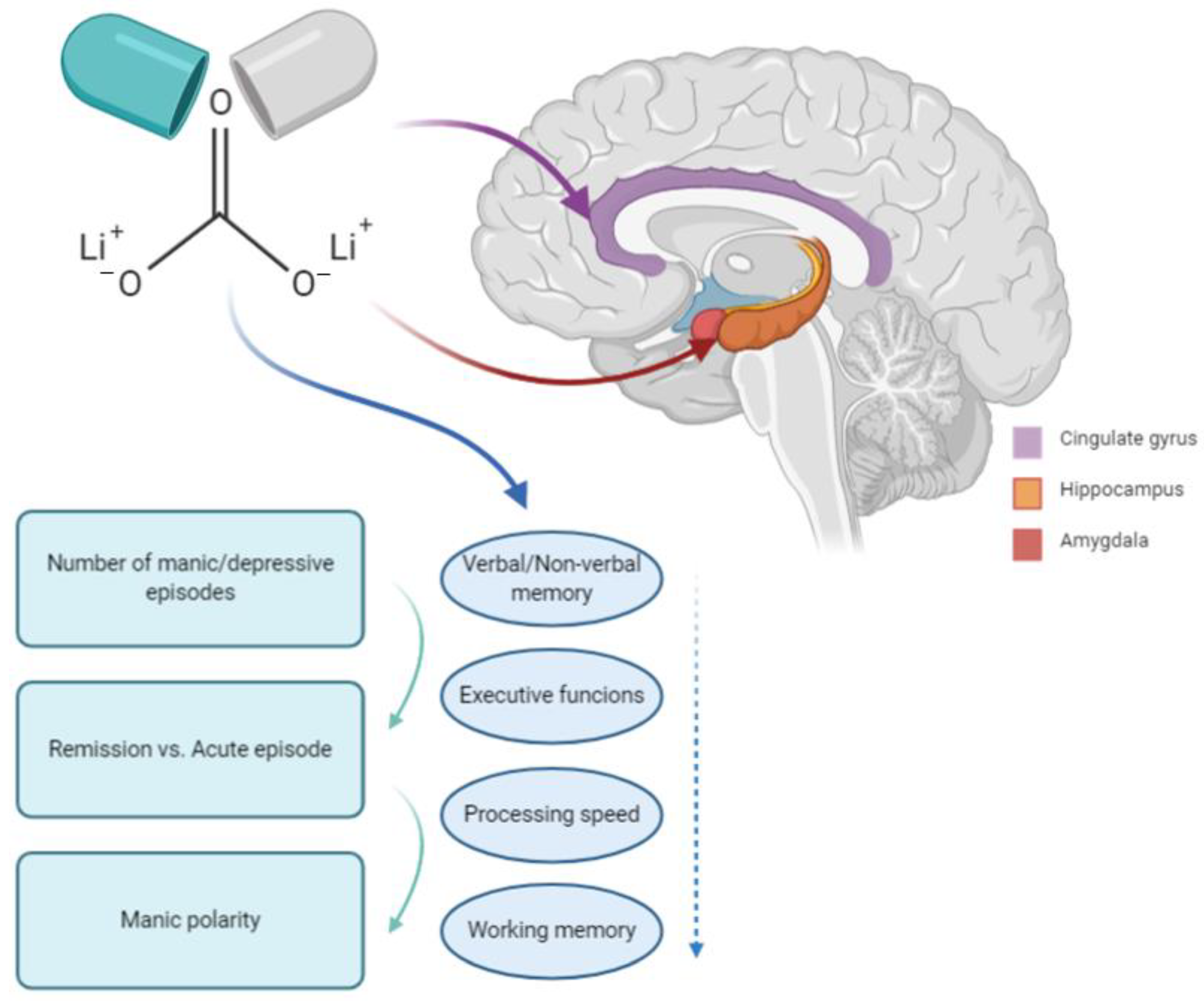{kind=link}
| Study | Study Design | Sample Size | Limitations | Conclusions |
|---|---|---|---|---|
| [25] | Cross-sectional | 30 euthymic type I BD patients; 30 HC | (i) Small sample size. (ii) 18/30 patients were on lithium medication, which could adversely affect cognitive performance. | Impairment of sustained attention may be considered as a neuropsychological marker for BD. |
| [26] | Cross-sectional | 25 euthymic, remitted type I BD patients; 25 HC | (i) Small sample size. (ii) 20/25 patients were on lithium medication. (iii) The effect of mood stabilizers on encoding strategies was not investigated. | The number of depressive and/or manic episodes, but not illness duration itself, was associated with impairments in organization abilities and non-verbal memory. |
| [24] | Cross-sectional | 40 euthymic BD patients; 30 HC | (i) Small sample size. (ii) Attention assessment was missing | A worse prior course of illness was related to poorer performance in delayed verbal memory and executive functions. |
| [28] | Cross-sectional | 45 newly diagnosed type I BD patients; 25 HC | (i) Both BD patients and HC had average or higher than average premorbid/current intellectual functioning. (ii) No correlation between cognitive performance and time elapsed between ratings and cognitive testing was found. | The neuropsychological deficit was present in clinically stable BD patients. |
| [43] | Longitudinal | 70 euthymic type I BD patients (49 with and 21 without significant cognitive impairment) | (i) All patients were taking psychotropic medications. (ii) Differences in impaired cognitive domains may not be equivalent in terms of the risk of recurrence. | Cognitive impairment might be considered the cause rather than the consequence of the clinical course. |
| [38] | Longitudinal | 57 BD patients; 31 HC | (i) Small sample size. (ii) Confounding effects of substance abuse/dependence. | Patients who were euthymic for one year after the illness onset had better cognitive performance compared to subjects with a recurrence of mood alterations. |
| [53] | Cross-sectional | 55 type I-II BD patients; 31 HC | (i) Small sample size of the subgroups. (ii) Influence of psychotropic medications. (iii) Exclusion of mixed episodes as polarity specifiers. | Manic polarity was associated with a greater cognitive impairment compared to BD with prevalent depressive polarity, regardless of the psychotic symptoms. |
| Study | Study Design | Sample Size | Technique | Limitations | Conclusions |
|---|---|---|---|---|---|
| [58] | Cross-sectional | 19 affective BD patients | CT | (i) HC sample was missing. (ii) Small sample size. (iii) Results only in subjects of 50 years of age or older. | A significant association between normalized ventricular volume and age was found. |
| [70] | Cross-sectional | 11 BD patients; 15 HC | 2.0 Tesla MRI | (i) Small sample size. | Reductions in fronto–temporo–limbic network regions were associated with poor clinical outcomes. |
| [65] | Longitudinal | 9 BD children; 8 HC | 1.5 Tesla MRI | (i) Small sample size. (ii) 6/9 patients were on lithium/mood stabilizers; (iii) All the children at the initial screening had psychotic features. | Increase in grey matter volume in the ventrolateral prefrontal cortex and temporal lobe structures. |
| [68] | Longitudinal | 20 type I BD patients; 21 HC | 1.5 Tesla MRI | (i) Small sample size. (ii) None of the participants were medically untreated. | The relative stability of the amygdala volume and hippocampal abnormalities was associated with poor cognitive outcomes over time in adults with BD. |
| [63] | Longitudinal | 10 type I BD patients; 8 HC | 1.5 Tesla MRI | (i) Small sample size. (ii) Wide age range (10–21 years old). (iii) Medication exposure. (iv) Variable interscan interval between groups. | The progressive prefrontal cortex volume loss was associated with disease severity in adolescents over two years. |
| [66] | Longitudinal | 58 BD patients; 48 HC | 4.0 Tesla MRI | (i) 48/58 patients were medically treated. (ii) Lack of assessment of the effect of illness course and duration, as well as medication status on structural changes. | The gray matter volume was increased in portions of the ventrolateral prefrontal cortex and hippocampus complex, centering on the parahippocampal gyrus but extending into the amygdala in BD patients. |
| [64] | Longitudinal | 16 BD patients; 70 HC | 1.5 Tesla MRI | (i) Small sample size. (ii) Analysis restricted to brain lobes. (iii) Patients had psychotic symptoms. | Progressive prefrontal cortex volume loss is associated with disease severity as assessed with the frequency of hospitalizations. |
| [71] | Cross-sectional | 125 type I BD patients; 305 HC | 1.5 Tesla MRI | (i) Usage of only one scale to measure impulsivity. (ii) The DSM-IV criteria rather than the DSM 5 criteria were used to categorize the participants. | Orbito–frontal abnormalities were associated with impulsivity and higher suicidal behavior. |
| Study | Study Design | Sample Size | Technique | Treatment | Limitations | Conclusions |
|---|---|---|---|---|---|---|
| [74] | Longitudinal | Animal model | / | Li2CO3 (4 eq/kg/day), sodium VPA (400 mg/dg/day), or saline by twice-daily intraperitoneal injections for 9 days or 4 weeks. | / | Lithium seems to be able to increase levels of the neuroprotective protein bcl-2 and promote neurite outgrowth. |
| [84] | Longitudinal | 12 untreated BD patients; 9 HC | 1.5 Tesla MRI | Four weeks of lithium administration (blinded) at therapeutic levels (0.8–l.2 meq/L). | (i) Small sample size. | The use of lithium was associated with an increase in a putative marker of neuronal integrity (N-acetyl-aspartate), both in HC and BD patients, providing in vivo evidence of its possible neurotropic effect. |
| [82,83] | Cross-sectional | 11 untreated BD patients; 16 BD patients treated with lithium; 39 HC | 1.5 Tesla MRI | Lithium monotherapy (for the lithium-treated group) | (i) Small sample size. (ii) Lack of a statistically significant correlation between cingulate volumes and specific clinical variables. (iii) The sample was composed of multiple-episode patients; thus, it is not clear whether the decreased anterior cingulate volume preceded the appearance of symptoms or appeared afterward. | Compared with untreated subjects, BD patients treated with lithium presented with increased grey matter volume in vivo, as seen using structural MRI-based whole-brain measures, and a decreased anterior cingulate volume. |
| [85] | Longitudinal | 12 BD patients | 1.5 Tesla MRI | Lithium | (i) Small sample size. (ii) Several subjects at the time of evaluation were treated with low dosages of other medications. (iii) The study did not contain a group of patients treated with medications other than lithium over the same period. | Progressively increased volume of the hippocampus after treatment with lithium, which was in association with improved verbal memory. |
| [87] | Cross-sectional + longitudinal | 262 BD patients (93 untreated, 169 lithium monotherapy) | / | Lithium | (i) Lack of a placebo control. (ii) Brief neurocognitive battery. (iii) Lack of cognitive testing available at a second time point in those patients who did not clinically respond to lithium. | Results from both cross-sectional and longitudinal analyses showed that lithium treatment did not significantly impair neurocognitive functions in patients with BD. |
| Study | Study Design | Sample Size | Technique | Limitations | Conclusions |
|---|---|---|---|---|---|
| [111] | Cross-sectional | 12 BD patients; 25 HC | 1.5 Tesla MRI; DTI | (i) Small sample size. | Fronto–striatal and temporal abnormalities have been observed in subjects with BD. |
| [107] | Cross-sectional | 32 euthymic type I BD patients; 30 HC | fMRI | (i) One-third ratio of unmedicated patients. (ii) Use of a block design, in which blocks of go-only events were contrasted against blocks composed of both go and no-go events presented randomly. (iii) The relationship between regional changes in basal ganglia and IFC could not be determined. (iv) The vulnerability for future episodes and activation of the IFC and striatal regions remain to be explored. | IFC, cingulate, and striatal structures were activated in BD patients while performing a response inhibition task. |
| [102] | Cross-sectional | 188 psychotic BD patients; 337 HC | MRI | (i) Confounding effect of medications. (ii) Imaging parameters were not optimized for imaging hippocampal subfields. | Widespread volumetric reductions in the hippocampus and its subfields within the spectrum of psychosis. |
| [101] | Cross-sectional | 117 type I BD patients, 66 type II BD patients; 300 HC | 1.5 Tesla MRI | (i) Possible confounding effects due to medication. (ii) The Bonferroni correction may be overly conservative because the hippocampal subfield volumes are not independent, and type II errors might have occurred. (iii) Using 1.5T MRI might have decreased the sensitivity to disease-related biological variability. | Smaller in vivo volumes of the hippocampal subfields CA2/3, CA4/DG, subiculum, and right CA1, along with smaller subiculum volume related to poorer immediate and delayed verbal recall in BD patients. |
| [98] | Longitudinal | 31 type I BD patients | 1.5 Tesla MRI | (i) Small sample size. (ii) Whole-brain analyses with a higher regional resolution, including the investigation of subcortical volumes, whereas larger study groups would give more detailed information on the effects of manic episodes on structural brain changes. iii) Lack of control data. iv) Probable presence of confounding factors given by differences in diet and exercise and undocumented drug use, as well as genetic, social, and environmental factors. | Progressive frontal cortical abnormalities were strongly related to manic episodes. Thirteen BD patients, labeled as the mania group, showed a decrease in cortical volume and area in both the dorso–lateral prefrontal cortex and inferior frontal cortex. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serafini, G.; Pardini, M.; Monacelli, F.; Orso, B.; Girtler, N.; Brugnolo, A.; Amore, M.; Nobili, F.; Team on Dementia of the IRCCS Ospedale Policlinico San Martino, D.M. Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature. Brain Sci. 2021, 11, 276. https://doi.org/10.3390/brainsci11020276
Serafini G, Pardini M, Monacelli F, Orso B, Girtler N, Brugnolo A, Amore M, Nobili F, Team on Dementia of the IRCCS Ospedale Policlinico San Martino DM. Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature. Brain Sciences. 2021; 11(2):276. https://doi.org/10.3390/brainsci11020276
Chicago/Turabian StyleSerafini, Gianluca, Matteo Pardini, Fiammetta Monacelli, Beatrice Orso, Nicola Girtler, Andrea Brugnolo, Mario Amore, Flavio Nobili, and Disease Management Team on Dementia of the IRCCS Ospedale Policlinico San Martino. 2021. "Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature" Brain Sciences 11, no. 2: 276. https://doi.org/10.3390/brainsci11020276
APA StyleSerafini, G., Pardini, M., Monacelli, F., Orso, B., Girtler, N., Brugnolo, A., Amore, M., Nobili, F., & Team on Dementia of the IRCCS Ospedale Policlinico San Martino, D. M. (2021). Neuroprogression as an Illness Trajectory in Bipolar Disorder: A Selective Review of the Current Literature. Brain Sciences, 11(2), 276. https://doi.org/10.3390/brainsci11020276