
Update on Paraneoplastic Cerebellar Degeneration

Abstract
1. Introduction
2. Principles of Autoimmunity
3. Epidemiology
4. Clinical Presentation
5. Evaluation
5.1. Diagnostic Criteria
5.2. Laboratory Testing
5.2.1. Antibodies Associated with Paraneoplastic Cerebellar Degeneration
5.2.2. Antibody Detection
5.2.3. Cerebrospinal Fluid Analysis
5.3. Imaging Studies
5.4. Cancer Search
5.5. Differential Diagnosis
6. Treatment and Management
6.1. Oncologic Treatments
6.2. Acute Immunotherapy
6.3. Maintenance Immunotherapy
7. Outcome and Prognosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitoma, H.; Manto, M.; Hadjivassiliou, M. Immune-mediated cerebellar ataxias: Clinical diagnosis and treatment based on immunological and physiological mechanisms. J. Mov. Disord. 2021, 14, 10–28. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Vogrig, A.; Gigli, G.L.; Segatti, S.; Corazza, E.; Marini, A.; Bernardini, A.; Valent, F.; Fabris, M.; Curcio, F.; Brigo, F.; et al. Epidemiology of paraneoplastic neurological syndromes: A population-based study. J. Neurol. 2020, 267, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, V. Beitrag zur kenntnis der chronishen diffusen kleinhirnerkrankungen. Neurol. Cbl. 1919, 38, 674–682. [Google Scholar]
- Greenlee, J.E.; Brashear, H.R. Antibodies to cerebellar purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann. Neurol. 1983, 14, 609–613. [Google Scholar] [CrossRef]
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.-C.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1014. [Google Scholar] [CrossRef]
- Loehrer, P.A.; Timmermann, L.; Pehl, A.; Bien, C.I.; Pfestroff, A.; Pedrosa, D.J. Rhombencephalitis associated with isolated zic4-antibodies in paraneoplastic cerebellar degeneration: A case report. BMC Neurol. 2020, 20, 208. [Google Scholar] [CrossRef]
- Höftberger, R.; Rosenfeld, M.R.; Dalmau, J. Update on neurological paraneoplastic syndromes. Curr. Opin. Oncol. 2015, 27, 489–495. [Google Scholar] [CrossRef]
- Pignolet, B.S.; Gebauer, C.M.; Liblau, R.S. Immunopathogenesis of paraneoplastic neurological syndromes associated with anti-hu antibodies: A beneficial antitumor immune response going awry. Oncoimmunology 2013, 2, e27384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qian, W.; Chen, Q.; Yin, L.; Li, B.; Wang, H. Imbalance in circulating t lymphocyte subsets contributes to hu antibody-associated paraneoplastic neurological syndromes. Cell. Immunol. 2014, 290, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Lang, B.; Pozo-Rosich, P.; Saiz, A.; Casamitjana, R.; Vincent, A. P/q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002, 59, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Lang, B.; Newsom-Davis, J. Autoimmunity to the voltage-gated calcium channel underlies the lambert-eaton myasthenic syndrome, a paraneoplastic disorder. Trends Neurosci. 1989, 12, 496–502. [Google Scholar] [CrossRef]
- Coesmans, M.; Smitt, P.A.; Linden, D.J.; Shigemoto, R.; Hirano, T.; Yamakawa, Y.; van Alphen, A.M.; Luo, C.; van der Geest, J.N.; Kros, J.M.; et al. Mechanisms underlying cerebellar motor deficits due to mglur1-autoantibodies. Ann. Neurol. 2003, 53, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Safa, P.; Chen, Y.R.; Sobel, R.A.; Boyden, E.S.; Tsien, R.W. Anti-Ca2+ channel antibody attenuates Ca2+ currents and mimics cerebellar ataxia in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2705–2710. [Google Scholar] [CrossRef]
- Peterson, K.; Rosenblum, M.K.; Kotanides, H.; Posner, J.B. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-yo antibody-positive patients. Neurology 1992, 42, 1931–1937. [Google Scholar] [CrossRef]
- Rojas, I.; Graus, F.; Keime-Guibert, F.; Reñé, R.; Delattre, J.Y.; Ramón, J.M.; Dalmau, J.; Posner, J.B. Long-term clinical outcome of paraneoplastic cerebellar degeneration and anti-yo antibodies. Neurology 2000, 55, 713–715. [Google Scholar] [CrossRef]
- Shams’ili, S.; Grefkens, J.; de Leeuw, B.; van den Bent, M.; Hooijkaas, H.; van der Holt, B.; Vecht, C.; Sillevis Smitt, P. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: Analysis of 50 patients. Brain J. Neurol. 2003, 126, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the cns. Lancet Neurol. 2008, 7, 327–340. [Google Scholar] [CrossRef]
- Grativvol, R.S.; Cavalcante, W.C.P.; Castro, L.H.M.; Nitrini, R.; Simabukuro, M.M. Updates in the diagnosis and treatment of paraneoplastic neurologic syndromes. Curr. Oncol. Rep. 2018, 20, 92. [Google Scholar] [CrossRef]
- Mitoma, H.; Adhikari, K.; Aeschlimann, D.; Chattopadhyay, P.; Hadjivassiliou, M.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Kakei, S.; Lee, J.; et al. Consensus paper: Neuroimmune mechanisms of cerebellar ataxias. Cerebellum 2016, 15, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Ducray, F.; Demarquay, G.; Graus, F.; Decullier, E.; Antoine, J.C.; Giometto, B.; Psimaras, D.; Delattre, J.Y.; Carpentier, A.F.; Honnorat, J. Seronegative paraneoplastic cerebellar degeneration: The pns euronetwork experience. Eur. J. Neurol. 2014, 21, 731–735. [Google Scholar] [CrossRef]
- Sechi, E.; Flanagan, E.P. Antibody-mediated autoimmune diseases of the cns: Challenges and approaches to diagnosis and management. Front. Neurol. 2021, 12, 673339. [Google Scholar] [CrossRef]
- Sillevis Smitt, P.A.; Manley, G.T.; Posner, J.B. Immunization with the paraneoplastic encephalomyelitis antigen hud does not cause neurologic disease in mice. Neurology 1995, 45, 1873–1878. [Google Scholar]
- Tanaka, K.; Tanaka, M.; Igarashi, S.; Onodera, O.; Miyatake, T.; Tsuji, S. Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-yo antibody. 2. Passive transfer of murine mononuclear cells activated with recombinant yo protein to paraneoplastic cerebellar degeneration lymphocytes in severe combined immunodeficiency mice. Clin. Neurol. Neurosurg. 1995, 97, 101–105. [Google Scholar]
- Carpentier, A.F.; Rosenfeld, M.R.; Delattre, J.Y.; Whalen, R.G.; Posner, J.B.; Dalmau, J. DNA vaccination with hud inhibits growth of a neuroblastoma in mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 2819–2824. [Google Scholar]
- Lancaster, E.; Dalmau, J. Neuronal autoantigens—Pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 2012, 8, 380–390. [Google Scholar] [CrossRef]
- Okano, H.J.; Park, W.Y.; Corradi, J.P.; Darnell, R.B. The cytoplasmic purkinje onconeural antigen cdr2 down-regulates c-myc function: Implications for neuronal and tumor cell survival. Genes Dev. 1999, 13, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kitagawa, Y.; Saiki, S.; Saiki, M.; Hirose, G. Effect of a paraneoplastic cerebellar degeneration-associated neural protein on b-myb promoter activity. Neurobiol. Dis. 2004, 15, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Austin, L.M.; Darnell, R.B. Detection and treatment of activated t cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann. Neurol. 2000, 47, 9–17. [Google Scholar] [CrossRef]
- Rousseau, A.; Benyahia, B.; Dalmau, J.; Connan, F.; Guillet, J.G.; Delattre, J.Y.; Choppin, J. T cell response to hu-d peptides in patients with anti-hu syndrome. J. Neuro-Oncol. 2005, 71, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Aye, M.M.; Kasai, T.; Tashiro, Y.; Xing, H.Q.; Shirahama, H.; Mitsuda, M.; Suetsugu, T.; Tanaka, K.; Osame, M.; Izumo, S. Cd8 positive t-cell infiltration in the dentate nucleus of paraneoplastic cerebellar degeneration. J. Neuroimmunol. 2009, 208, 136–140. [Google Scholar] [CrossRef]
- Plonquet, A.; Gherardi, R.K.; Créange, A.; Antoine, J.C.; Benyahia, B.; Grisold, W.; Drlicek, M.; Dreyfus, P.; Honnorat, J.; Khouatra, C.; et al. Oligoclonal t-cells in blood and target tissues of patients with anti-hu syndrome. J. Neuroimmunol. 2002, 122, 100–105. [Google Scholar] [CrossRef]
- Voltz, R.; Dalmau, J.; Posner, J.B.; Rosenfeld, M.R. T-cell receptor analysis in anti-hu associated paraneoplastic encephalomyelitis. Neurology 1998, 51, 1146–1150. [Google Scholar] [CrossRef]
- Fukuda, T.; Motomura, M.; Nakao, Y.; Shiraishi, H.; Yoshimura, T.; Iwanaga, K.; Tsujihata, M.; Eguchi, K. Reduction of p/q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with lambert-eaton myasthenic syndrome. Ann. Neurol. 2003, 53, 21–28. [Google Scholar] [CrossRef]
- Hildebrand, M.E.; Isope, P.; Miyazaki, T.; Nakaya, T.; Garcia, E.; Feltz, A.; Schneider, T.; Hescheler, J.; Kano, M.; Sakimura, K.; et al. Functional coupling between mglur1 and cav3.1 t-type calcium channels contributes to parallel fiber-induced fast calcium signaling within purkinje cell dendritic spines. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9668–9682. [Google Scholar] [CrossRef]
- Hébert, J.; Riche, B.; Vogrig, A.; Muñiz-Castrillo, S.; Joubert, B.; Picard, G.; Rogemond, V.; Psimaras, D.; Alentorn, A.; Berzero, G.; et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in france. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e883. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kwon, S.; Ki, C.S.; Youn, J.; Cho, J.W. The etiologies of chronic progressive cerebellar ataxia in a korean population. J. Clin. Neurol. 2018, 14, 374–380. [Google Scholar] [CrossRef]
- Gebus, O.; Montaut, S.; Monga, B.; Wirth, T.; Cheraud, C.; Alves Do Rego, C.; Zinchenko, I.; Carré, G.; Hamdaoui, M.; Hautecloque, G.; et al. Deciphering the causes of sporadic late-onset cerebellar ataxias: A prospective study with implications for diagnostic work. J. Neurol. 2017, 264, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- de Graaff, E.; Maat, P.; Hulsenboom, E.; van den Berg, R.; van den Bent, M.; Demmers, J.; Lugtenburg, P.J.; Hoogenraad, C.C.; Sillevis Smitt, P. Identification of delta/notch-like epidermal growth factor-related receptor as the tr antigen in paraneoplastic cerebellar degeneration. Ann. Neurol. 2012, 71, 815–824. [Google Scholar] [CrossRef]
- McKeon, A.; Tracy, J.A.; Pittock, S.J.; Parisi, J.E.; Klein, C.J.; Lennon, V.A. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: The cerebellum and beyond. Arch. Neurol. 2011, 68, 1282–1289. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F.; Rosenblum, M.K.; Posner, J.B. Anti-hu--associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine 1992, 71, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A. Anti-neuronal nuclear autoantibody type 2: Paraneoplastic accompaniments. Ann. Neurol. 2003, 53, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K.; Wick, M.; Roth, G.; Decker, P.; Voltz, R. Antineuronal antibodies in sporadic late-onset cerebellar ataxia. J. Neurol. 2010, 257, 59–62. [Google Scholar] [CrossRef]
- Sabater, L.; Höftberger, R.; Boronat, A.; Saiz, A.; Dalmau, J.; Graus, F. Antibody repertoire in paraneoplastic cerebellar degeneration and small cell lung cancer. PLoS ONE 2013, 8, e60438. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Delattre, J.Y.; Antoine, J.C.; Dalmau, J.; Giometto, B.; Grisold, W.; Honnorat, J.; Smitt, P.S.; Vedeler, C.; Verschuuren, J.J.; et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Lennon, V.A.; Gadoth, A.; Pittock, S.J.; Flanagan, E.P.; Schmeling, J.E.; McKeon, A.; Klein, C.J. Autoimmune crmp5 neuropathy phenotype and outcome defined from 105 cases. Neurology 2018, 90, e103–e110. [Google Scholar] [CrossRef] [PubMed]
- Honnorat, J.; Cartalat-Carel, S.; Ricard, D.; Camdessanche, J.P.; Carpentier, A.F.; Rogemond, V.; Chapuis, F.; Aguera, M.; Decullier, E.; Duchemin, A.M.; et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with hu or cv2/crmp5 antibodies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 412–416. [Google Scholar] [CrossRef]
- Yu, Z.; Kryzer, T.J.; Griesmann, G.E.; Kim, K.; Benarroch, E.E.; Lennon, V.A. Crmp-5 neuronal autoantibody: Marker of lung cancer and thymoma-related autoimmunity. Ann. Neurol. 2001, 49, 146–154. [Google Scholar] [CrossRef]
- Graus, F.; Keime-Guibert, F.; Rene, R.; Benyahia, B.; Ribalta, T.; Ascaso, C.; Escaramis, G.; Delattre, J.Y. Anti-hu-associated paraneoplastic encephalomyelitis: Analysis of 200 patients. Brain J. Neurol. 2001, 124, 1138–1148. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F.; Villarejo, A.; Posner, J.B.; Blumenthal, D.; Thiessen, B.; Saiz, A.; Meneses, P.; Rosenfeld, M.R. Clinical analysis of anti-ma2-associated encephalitis. Brain J. Neurol. 2004, 127, 1831–1844. [Google Scholar] [CrossRef]
- Hoffmann, L.A.; Jarius, S.; Pellkofer, H.L.; Schueller, M.; Krumbholz, M.; Koenig, F.; Johannis, W.; la Fougere, C.; Newman, T.; Vincent, A.; et al. Anti-ma and anti-ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J. Neurol. Neurosurg. Psychiatry 2008, 79, 767–773. [Google Scholar] [CrossRef]
- Simard, C.; Vogrig, A.; Joubert, B.; Muñiz-Castrillo, S.; Picard, G.; Rogemond, V.; Ducray, F.; Berzero, G.; Psimaras, D.; Antoine, J.C.; et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with ri antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e699. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.; Shams’ili, S.; Rojas, I.; Sanchez-Valle, R.; Saiz, A.; Dalmau, J.; Honnorat, J.; Sillevis Smitt, P.; Graus, F. Anti-tr antibodies as markers of paraneoplastic cerebellar degeneration and hodgkin’s disease. Neurology 2003, 60, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Jitprapaikulsan, J.; Bi, H.; Do Campo, R.V.; McKeon, A.; Pittock, S.J.; Engelstad, J.K.; Mills, J.R.; Klein, C.J. Amphiphysin-igg autoimmune neuropathy: A recognizable clinicopathologic syndrome. Neurology 2019, 93, e1873–e1880. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Lucchinetti, C.F.; Parisi, J.E.; Benarroch, E.E.; Mokri, B.; Stephan, C.L.; Kim, K.K.; Kilimann, M.W.; Lennon, V.A. Amphiphysin autoimmunity: Paraneoplastic accompaniments. Ann. Neurol. 2005, 58, 96–107. [Google Scholar] [CrossRef]
- Dubey, D.; Hinson, S.R.; Jolliffe, E.A.; Zekeridou, A.; Flanagan, E.P.; Pittock, S.J.; Basal, E.; Drubach, D.A.; Lachance, D.H.; Lennon, V.A.; et al. Autoimmune gfap astrocytopathy: Prospective evaluation of 90 patients in 1 year. J. Neuroimmunol. 2018, 321, 157–163. [Google Scholar] [CrossRef]
- Saiz, A.; Blanco, Y.; Sabater, L.; González, F.; Bataller, L.; Casamitjana, R.; Ramió-Torrentà, L.; Graus, F. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: Diagnostic clues for this association. Brain J. Neurol. 2008, 131, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Mandel-Brehm, C.; Dubey, D.; Kryzer, T.J.; O’Donovan, B.D.; Tran, B.; Vazquez, S.E.; Sample, H.A.; Zorn, K.C.; Khan, L.M.; Bledsoe, I.O.; et al. Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis. N. Engl. J. Med. 2019, 381, 47–54. [Google Scholar] [CrossRef]
- Maudes, E.; Landa, J.; Muñoz-Lopetegi, A.; Armangue, T.; Alba, M.; Saiz, A.; Graus, F.; Dalmau, J.; Sabater, L. Clinical significance of kelch-like protein 11 antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e666. [Google Scholar] [CrossRef]
- Dubey, D.; Wilson, M.R.; Clarkson, B.; Giannini, C.; Gandhi, M.; Cheville, J.; Lennon, V.A.; Eggers, S.; Devine, M.F.; Mandel-Brehm, C.; et al. Expanded clinical phenotype, oncological associations, and immunopathologic insights of paraneoplastic kelch-like protein-11 encephalitis. JAMA Neurol. 2020, 77, 1420–1429. [Google Scholar] [CrossRef]
- Zis, P.; Rao, D.G.; Hoggard, N.; Sarrigiannis, P.G.; Hadjivassiliou, M. Anti-mag associated cerebellar ataxia and response to rituximab. J. Neurol. 2018, 265, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Vernino, S.; Lennon, V.A. New purkinje cell antibody (pca-2): Marker of lung cancer-related neurological autoimmunity. Ann. Neurol. 2000, 47, 297–305. [Google Scholar] [CrossRef]
- Gadoth, A.; Kryzer, T.J.; Fryer, J.; McKeon, A.; Lennon, V.A.; Pittock, S.J. Microtubule-associated protein 1b: Novel paraneoplastic biomarker. Ann. Neurol. 2017, 81, 266–277. [Google Scholar] [CrossRef]
- Irani, S.R.; Pettingill, P.; Kleopa, K.A.; Schiza, N.; Waters, P.; Mazia, C.; Zuliani, L.; Watanabe, O.; Lang, B.; Buckley, C.; et al. Morvan syndrome: Clinical and serological observations in 29 cases. Ann. Neurol. 2012, 72, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-Castrillo, S.; Joubert, B.; Elsensohn, M.H.; Pinto, A.L.; Saint-Martin, M.; Vogrig, A.; Picard, G.; Rogemond, V.; Dubois, V.; Tamouza, R.; et al. Anti-caspr2 clinical phenotypes correlate with hla and immunological features. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1076–1084. [Google Scholar] [CrossRef]
- Joubert, B.; Saint-Martin, M.; Noraz, N.; Picard, G.; Rogemond, V.; Ducray, F.; Desestret, V.; Psimaras, D.; Delattre, J.Y.; Antoine, J.C.; et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. 2016, 73, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. Dppx potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef]
- Hara, M.; Ariño, H.; Petit-Pedrol, M.; Sabater, L.; Titulaer, M.J.; Martinez-Hernandez, E.; Schreurs, M.W.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. Dppx antibody-associated encephalitis: Main syndrome and antibody effects. Neurology 2017, 88, 1340–1348. [Google Scholar] [CrossRef]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, morvan’s syndrome and acquired neuromyotonia. Brain J. Neurol. 2010, 133, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Gadoth, A.; Pittock, S.J.; Dubey, D.; McKeon, A.; Britton, J.W.; Schmeling, J.E.; Smith, A.; Kotsenas, A.L.; Watson, R.E.; Lachance, D.H.; et al. Expanded phenotypes and outcomes among 256 lgi1/caspr2-igg-positive patients. Ann. Neurol. 2017, 82, 79–92. [Google Scholar] [CrossRef]
- Lopez-Chiriboga, A.S.; Komorowski, L.; Kümpfel, T.; Probst, C.; Hinson, S.R.; Pittock, S.J.; McKeon, A. Metabotropic glutamate receptor type 1 autoimmunity: Clinical features and treatment outcomes. Neurology 2016, 86, 1009–1013. [Google Scholar] [CrossRef]
- Zalewski, N.L.; Lennon, V.A.; Lachance, D.H.; Klein, C.J.; Pittock, S.J.; McKeon, A. P/q- and n-type calcium-channel antibodies: Oncological, neurological, and serological accompaniments. Muscle Nerve 2016, 54, 220–227. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Graus, F.; Honnorat, J.; Jarius, S.; Titulaer, M.; Manto, M.; Hoggard, N.; Sarrigiannis, P.; Mitoma, H. Diagnostic criteria for primary autoimmune cerebellar ataxia-guidelines from an international task force on immune-mediated cerebellar ataxias. Cerebellum 2020, 19, 605–610. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B. ‘Medusa head ataxia’: The expanding spectrum of purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-pkc-gamma, anti-glur-delta2, anti-ca/arhgap26 and anti-vgcc. J. Neuroinflamm. 2015, 12, 167. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Alfugham, N.; O’Connor, K.; Hinson, S.; Kunchok, A.; Lennon, V.A.; Komorowski, L.; Probst, C.; McKeon, A. Gtpase regulator associated with focal adhesion kinase 1 (graf1) immunoglobulin-associated ataxia and neuropathy. Mov. Disord. Clin. Pract. 2020, 7, 904–909. [Google Scholar] [CrossRef]
- Bataller, L.; Sabater, L.; Saiz, A.; Serra, C.; Claramonte, B.; Graus, F. Carbonic anhydrase-related protein viii: Autoantigen in paraneoplastic cerebellar degeneration. Ann. Neurol. 2004, 56, 575–579. [Google Scholar] [CrossRef]
- Höftberger, R.; Sabater, L.; Velasco, F.; Ciordia, R.; Dalmau, J.; Graus, F. Carbonic anhydrase-related protein viii antibodies and paraneoplastic cerebellar degeneration. Neuropathol. Appl. Neurobiol. 2014, 40, 650–653. [Google Scholar] [CrossRef]
- Prevezianou, A.; Tzartos, J.S.; Dagklis, I.E.; Bentenidi, E.; Angelopoulos, P.; Bostantjopoulou, S. Paraneoplastic cerebellar degeneration in a patient with breast cancer associated with carbonic anhydrase-related protein viii autoantibodies. J. Neuroimmunol. 2020, 344, 577242. [Google Scholar] [CrossRef] [PubMed]
- Swayne, A.; Tjoa, L.; Broadley, S.; Dionisio, S.; Gillis, D.; Jacobson, L.; Woodhall, M.R.; McNabb, A.; Schweitzer, D.; Tsang, B.; et al. Antiglycine receptor antibody related disease: A case series and literature review. Eur. J. Neurol. 2018, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. ‘Medusa-head ataxia’: The expanding spectrum of purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: Anti-mglur1, anti-homer-3, anti-sj/itpr1 and anti-carp viii. J. Neuroinflamm. 2015, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ren, H.; Li, L.; Wang, J.; Fechner, K.; Guan, H. Anti-homer-3 antibody associated cerebellar ataxia: A rare case report and literature review. J. Neuroimmunol. 2019, 330, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, L.; Sabater, L.; Saiz, A.; Baiges, J.J.; Giometto, B.; Graus, F. Homer 3 autoimmunity in subacute idiopathic cerebellar ataxia. Neurology 2007, 68, 239–240. [Google Scholar] [CrossRef]
- Höftberger, R.; Sabater, L.; Ortega, A.; Dalmau, J.; Graus, F. Patient with homer-3 antibodies and cerebellitis. JAMA Neurol. 2013, 70, 506–509. [Google Scholar] [CrossRef]
- Ruiz-García, R.; Martínez-Hernández, E.; Joubert, B.; Petit-Pedrol, M.; Pajarón-Boix, E.; Fernández, V.; Salais, L.; Del Pozo, M.; Armangué, T.; Sabater, L.; et al. Paraneoplastic cerebellar ataxia and antibodies to metabotropic glutamate receptor 2. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e658. [Google Scholar] [CrossRef]
- Mange, L.; Haitao, R.; Lixin, Z.; Siyuan, F.; Jing, W.; Hongzhi, G. Cerebellar ataxia and myeloradiculopathy associated with ap3b2 antibody: A case report and literature review. J. Neurol. 2021, 268, 4163–4169. [Google Scholar] [CrossRef]
- Darnell, R.B.; Furneaux, H.M.; Posner, J.B. Antiserum from a patient with cerebellar degeneration identifies a novel protein in purkinje cells, cortical neurons, and neuroectodermal tumors. J. Neurosci. Off. J. Soc. Neurosci. 1991, 11, 1224–1230. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B. ‘Medusa head ataxia’: The expanding spectrum of purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: Anti-yo/cdr2, anti-nb/ap3b2, pca-2, anti-tr/dner, other antibodies, diagnostic pitfalls, summary and outlook. J. Neuroinflamm. 2015, 12, 168. [Google Scholar] [CrossRef]
- Miske, R.; Gross, C.C.; Scharf, M.; Golombeck, K.S.; Hartwig, M.; Bhatia, U.; Schulte-Mecklenbeck, A.; Bönte, K.; Strippel, C.; Schöls, L.; et al. Neurochondrin is a neuronal target antigen in autoimmune cerebellar degeneration. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e307. [Google Scholar] [CrossRef]
- Shelly, S.; Kryzer, T.J.; Komorowski, L.; Miske, R.; Anderson, M.D.; Flanagan, E.P.; Hinson, S.R.; Lennon, V.A.; Pittock, S.J.; McKeon, A. Neurochondrin neurological autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ren, H.; Fang, F.; Yang, X.; Wang, J.; Guan, H. Neurochondrin antibody serum positivity in three cases of autoimmune cerebellar ataxia. Cerebellum 2019, 18, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Shelly, S.; Zivelonghi, C.; Basal, E.; Dubey, D.; Flanagan, E.; Madhavan, A.A.; Mariotto, S.; Toledano, M.; Tracy, J.A.; et al. Neuronal intermediate filament iggs in csf: Autoimmune axonopathy biomarkers. Ann. Clin. Transl. Neurol. 2021, 8, 425–439. [Google Scholar] [CrossRef]
- Sabater, L.; Bataller, L.; Carpentier, A.F.; Aguirre-Cruz, M.L.; Saiz, A.; Benyahia, B.; Dalmau, J.; Graus, F. Protein kinase cgamma autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, R.; Kovacs, G.G.; Sabater, L.; Nagy, P.; Racz, G.; Miquel, R.; Dalmau, J.; Graus, F. Protein kinase cγ antibodies and paraneoplastic cerebellar degeneration. J. Neuroimmunol. 2013, 256, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Honorat, J.A.; Lopez-Chiriboga, A.S.; Kryzer, T.J.; Fryer, J.P.; Devine, M.; Flores, A.; Lennon, V.A.; Pittock, S.J.; McKeon, A. Autoimmune septin-5 cerebellar ataxia. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e474. [Google Scholar] [CrossRef]
- Yaguchi, H.; Yabe, I.; Takahashi, H.; Okumura, F.; Takeuchi, A.; Horiuchi, K.; Kano, T.; Kanda, A.; Saito, W.; Matsumoto, M.; et al. Identification of anti-sez6l2 antibody in a patient with cerebellar ataxia and retinopathy. J. Neurol. 2014, 261, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Hahn, S.; Hanssen, H.; Münchau, A.; Wandinger, K.-P.; Brüggemann, N. Sez6l2-antibody-associated progressive cerebellar ataxia: A differential diagnosis of atypical parkinsonism. J. Neurol. 2019, 266, 522–524. [Google Scholar] [CrossRef]
- Landa, J.; Guasp, M.; Petit-Pedrol, M.; Martínez-Hernández, E.; Planagumà, J.; Saiz, A.; Ruiz-García, R.; García-Fernández, L.; Verschuuren, J.; Saunders-Pullman, R.; et al. Seizure-related 6 homolog like 2 autoimmunity: Neurologic syndrome and antibody effects. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, 213–232. [Google Scholar] [CrossRef]
- Jarius, S.; Ringelstein, M.; Haas, J.; Serysheva, I.I.; Komorowski, L.; Fechner, K.; Wandinger, K.-P.; Albrecht, P.; Hefter, H.; Moser, A.; et al. Inositol 1,4,5-trisphosphate receptor type 1 autoantibodies in paraneoplastic and non-paraneoplastic peripheral neuropathy. J. Neuroinflamm. 2016, 13, 278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jarius, S.; Scharf, M.; Begemann, N.; Stöcker, W.; Probst, C.; Serysheva, I.I.; Nagel, S.; Graus, F.; Psimaras, D.; Wildemann, B.; et al. Antibodies to the inositol 1,4,5-trisphosphate receptor type 1 (itpr1) in cerebellar ataxia. J. Neuroinflamm. 2014, 11, 206. [Google Scholar] [CrossRef]
- Berzero, G.; Hacohen, Y.; Komorowski, L.; Scharf, M.; Dehais, C.; Leclercq, D.; Fourchotte, V.; Buecher, B.; Honnorat, J.; Graus, F.; et al. Paraneoplastic cerebellar degeneration associated with anti-itpr1 antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e326. [Google Scholar] [CrossRef]
- Alfugham, N.; Gadoth, A.; Lennon, V.A.; Komorowski, L.; Scharf, M.; Hinson, S.; McKeon, A.; Pittock, S.J. Itpr1 Autoimmunity: Frequency, Neurologic Phenotype, and Cancer Association; Rehobot Atamiwoc: Addis Ababa, Ethiopia, 2018; 294p. [Google Scholar]
- Sun, X.; Tan, J.; Sun, H.; Liu, Y.; Guan, W.; Jia, J.; Wang, Z. Anti-sox1 antibodies in paraneoplastic neurological syndrome. J. Clin. Neurol. 2020, 16, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Le Do, D.; Gupton, S.L.; Tanji, K.; Bastien, J.; Brugière, S.; Couté, Y.; Quadrio, I.; Rogemond, V.; Fabien, N.; Desestret, V.; et al. Trim9 and trim67 are new targets in paraneoplastic cerebellar degeneration. Cerebellum 2019, 18, 245–254. [Google Scholar]
- van Coevorden-Hameete, M.H.; van Beuningen, S.F.B.; Perrenoud, M.; Will, L.M.; Hulsenboom, E.; Demonet, J.-F.; Sabater, L.; Kros, J.M.; Verschuuren, J.J.G.M.; Titulaer, M.J.; et al. Antibodies to trim46 are associated with paraneoplastic neurological syndromes. Ann. Clin. Transl. Neurol. 2017, 4, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Larman, H.B.; Zhao, Z.; Laserson, U.; Li, M.Z.; Ciccia, A.; Gakidis, M.A.M.; Church, G.M.; Kesari, S.; Leproust, E.M.; Solimini, N.L.; et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011, 29, 535–541. [Google Scholar] [CrossRef]
- Etemadifar, M.; Aghababaei, A.; Nouri, H.; Kargaran, P.K.; Mohammadi, S.; Salari, M. Autoimmune encephalitis: The first observational study from iran. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 1–10. [Google Scholar] [CrossRef]
- Iyer, S.G.; Khakoo, N.S.; Aitcheson, G.; Perez, C. Case of anti-zic4 antibody-mediated cerebellar toxicity induced by dual checkpoint inhibition in head and neck squamous cell carcinoma. BMJ Case Rep. 2020, 13, e235607. [Google Scholar] [CrossRef]
- Gresa-Arribas, N.; Planaguma, J.; Petit-Pedrol, M.; Kawachi, I.; Katada, S.; Glaser, C.A.; Simabukuro, M.M.; Armangue, T.; Martinez-Hernandez, E.; Graus, F.; et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology 2016, 86, 2235–2242. [Google Scholar] [CrossRef]
- Loehrer, P.A.; Bien, C.I.; Dusoi, A.E.; Timmermann, L.; Simon, O.J. Neurexin-3α-associated autoimmune encephalitis: A case report of full recovery after rituximab therapy. Eur. J. Neurol. 2020, 27, e91–e93. [Google Scholar] [CrossRef] [PubMed]
- Inui, R.; Saito, K.; Shimomura, Y.; Yamashita, D.; Kawamoto, M.; Ishikawa, T. Anti-ma-associated paraneoplastic cerebellar degeneration in a patient with nodular lymphocyte-predominant hodgkin lymphoma: A case report. BMC Neurol. 2020, 20, 355. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Xu, S.-H.; Zhang, S.-R.; Shu, Q.-F.; Liu, X.-L. Anti-yo antibody-positive paraneoplastic cerebellar degeneration in a patient with possible cholangiocarcinoma: A case report and review of the literature. World J. Clin. Cases 2021, 9, 4423–4432. [Google Scholar] [CrossRef]
- Escudero, D.; Guasp, M.; Ariño, H.; Gaig, C.; Martínez-Hernández, E.; Dalmau, J.; Graus, F. Antibody-associated cns syndromes without signs of inflammation in the elderly. Neurology 2017, 89, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Saiz, A.; Graus, F.; Dalmau, J.; Pifarré, A.; Marín, C.; Tolosa, E. Detection of 14-3-3 brain protein in the cerebrospinal fluid of patients with paraneoplastic neurological disorders. Ann. Neurol. 1999, 46, 774–777. [Google Scholar] [CrossRef]
- Mascalchi, M.; Vella, A. Magnetic resonance and nuclear medicine imaging in ataxias. Handb. Clin. Neurol. 2012, 103, 85–110. [Google Scholar] [PubMed]
- Baldarçara, L.; Currie, S.; Hadjivassiliou, M.; Hoggard, N.; Jack, A.; Jackowski, A.P.; Mascalchi, M.; Parazzini, C.; Reetz, K.; Righini, A.; et al. Consensus paper: Radiological biomarkers of cerebellar diseases. Cerebellum 2015, 14, 175–196. [Google Scholar] [CrossRef]
- Madhavan, A.A.; Carr, C.M.; Morris, P.P.; Flanagan, E.P.; Kotsenas, A.L.; Hunt, C.H.; Eckel, L.J.; Lindell, E.P.; Diehn, F.E. Imaging review of paraneoplastic neurologic syndromes. AJNR. Am. J. Neuroradiol. 2020, 41, 2176–2187. [Google Scholar] [CrossRef] [PubMed]
- Mahta, A.; Vijayvergia, N.; Bhavsar, T.M.; Ward, L.D. Diagnostic approach to a patient with paraneoplastic neurological syndrome. World J. Oncol. 2012, 3, 243–246. [Google Scholar] [CrossRef]
- Gilmore, C.P.; Elliott, I.; Auer, D.; Maddison, P. Diffuse cerebellar mr imaging changes in anti-yo positive paraneoplastic cerebellar degeneration. J. Neurol. 2010, 257, 490–491. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Komatsu, K.; Nakagawa, T.; Matsumoto, S. Transient diffusion-weighted imaging hyperintensity of the cerebellar cortex in paraneoplastic cerebellar degeneration. Intern. Med. 2019, 58, 619–620. [Google Scholar] [CrossRef]
- Karmon, Y.; Inbar, E.; Cordoba, M.; Gadoth, N. Paraneoplastic cerebellar degeneration mimicking acute post-infectious cerebellitis. Cerebellum 2009, 8, 441–444. [Google Scholar] [CrossRef]
- Choi, K.-D.; Kim, J.S.; Park, S.-H.; Kim, Y.K.; Kim, S.E.; Smitt, P.S. Cerebellar hypermetabolism in paraneoplastic cerebellar degeneration. J. Neurol. Neurosurg. Psychiatry 2006, 77, 525–528. [Google Scholar] [CrossRef]
- Kroiss, A.; Uprimny, C.; Virgolini, I.J. Thyroid carcinoma detected by incidental (18)f-fdg uptake in a patient with progressive cerebellar syndrome. Endocrine 2016, 51, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Zhao, Y.; Cui, R. Cerebellar hypermetabolism in a case of paraneoplastic cerebellar syndrome with the primary lymphoepithelial carcinoma in tonsil. Clin. Nucl. Med. 2019, 44, 812–814. [Google Scholar] [CrossRef]
- Titulaer, M.J.; Soffietti, R.; Dalmau, J.; Gilhus, N.E.; Giometto, B.; Graus, F.; Grisold, W.; Honnorat, J.; Sillevis Smitt, P.A.E.; Tanasescu, R.; et al. Screening for tumours in paraneoplastic syndromes: Report of an efns task force. Eur. J. Neurol. 2011, 18, 19-e13. [Google Scholar] [CrossRef]
- Ropper, A.H.; Samuels, M.A.; Klein, P.J. Adams and Victor’s Principles of Neurology; McGraw-Hill Medical: New York, NY, USA, 2019. [Google Scholar]
- Mitoma, H.; Hadjivassiliou, M.; Honnorat, J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015, 2, 14. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Dalmau, J. Update on paraneoplastic and autoimmune disorders of the central nervous system. Semin. Neurol. 2010, 30, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-nmda receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Witt, K.A.; Sandoval, K.E. Steroids and the blood-brain barrier: Therapeutic implications. Adv. Pharmacol. 2014, 71, 361–390. [Google Scholar]
- Shibata, T.; Oishi, T.; Fukuoka, Y.; Nishikawa, S.; Iizuka, N.; Kato, H. Potential effect of intravenous immunoglobulin against paraneoplastic cerebellar degeneration in progressive ovarian cancer. Gynecol. Oncol. Rep. 2020, 34, 100649. [Google Scholar] [CrossRef]
- Hu, F.Q.; Shang, F.R.; Liu, J.J.; Yuan, H. Plasma exchange for treating anti-yo-associated paraneoplastic cerebellar degeneration: Case report and literature review. Medicine 2020, 99, e21760. [Google Scholar] [CrossRef]
- Guo, Y.; Tian, X.; Wang, X.; Xiao, Z. Adverse effects of immunoglobulin therapy. Front. Immunol. 2018, 9, 1299. [Google Scholar] [CrossRef]
- Flabeau, O.; Laurent, C.; Schneider, S.; Honnorat, J.; Ellie, E. Spinal cord tractopathy in paraneoplastic anti-cv2/crmp5 myelitis responsive to plasma exchange. Rev. Neurol. 2021. [Google Scholar] [CrossRef]
- Mizenko, C.; Bennett, J.L.; Owens, G.; Vollmer, T.L.; Piquet, A.L. A longitudinal, observational analysis of neuronal injury biomarkers in a case report of a patient with paraneoplastic anti-crmp5 antibody-associated transverse myelitis. Front. Neurol. 2021, 12, 691509. [Google Scholar] [CrossRef] [PubMed]
- Chevret, S.; Hughes, R.A.; Annane, D. Plasma exchange for guillain-barré syndrome. Cochrane Database Syst. Rev. 2017, 2, Cd001798. [Google Scholar] [CrossRef]
- Rath, J.; Zulehner, G.; Schober, B.; Grisold, A.; Krenn, M.; Cetin, H.; Zimprich, F. Real-world treatment of adult patients with guillain-barré syndrome over the last two decades. Sci. Rep. 2021, 11, 19170. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.; Probasco, J.C.; Irani, S.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune encephalitis: Proposed best practice recommendations for diagnosis and acute management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Grunewald, R.A.; Shanmugarajah, P.D.; Sarrigiannis, P.G.; Zis, P.; Skarlatou, V.; Hoggard, N. Treatment of primary autoimmune cerebellar ataxia with mycophenolate. Cerebellum 2020, 19, 680–684. [Google Scholar] [CrossRef]
- Esposito, M.; Penza, P.; Orefice, G.; Pagano, A.; Parente, E.; Abbadessa, A.; Bonavita, V. Successful treatment of paraneoplastic cerebellar degeneration with rituximab. J. Neuro-Oncol. 2008, 86, 363–364. [Google Scholar] [CrossRef]
- Poepel, A.; Jarius, S.; Heukamp, L.C.; Urbach, H.; Elger, C.E.; Bien, C.G.; Voltz, R. Neurological course of long-term surviving patients with sclc and anti-hu syndrome. J. Neurol. Sci. 2007, 263, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Voltz, R. Paraneoplastic neurological syndromes: An update on diagnosis, pathogenesis, and therapy. Lancet Neurol. 2002, 1, 294–305. [Google Scholar] [CrossRef]
- Bataller, L.; Graus, F.; Saiz, A.; Vilchez, J.J. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain J. Neurol. 2001, 124, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Stark, E.; Wurster, U.; Patzold, U.; Sailer, M.; Haas, J. Immunological and clinical response to immunosuppressive treatment in paraneoplastic cerebellar degeneration. Arch. Neurol. 1995, 52, 814–818. [Google Scholar] [CrossRef] [PubMed]


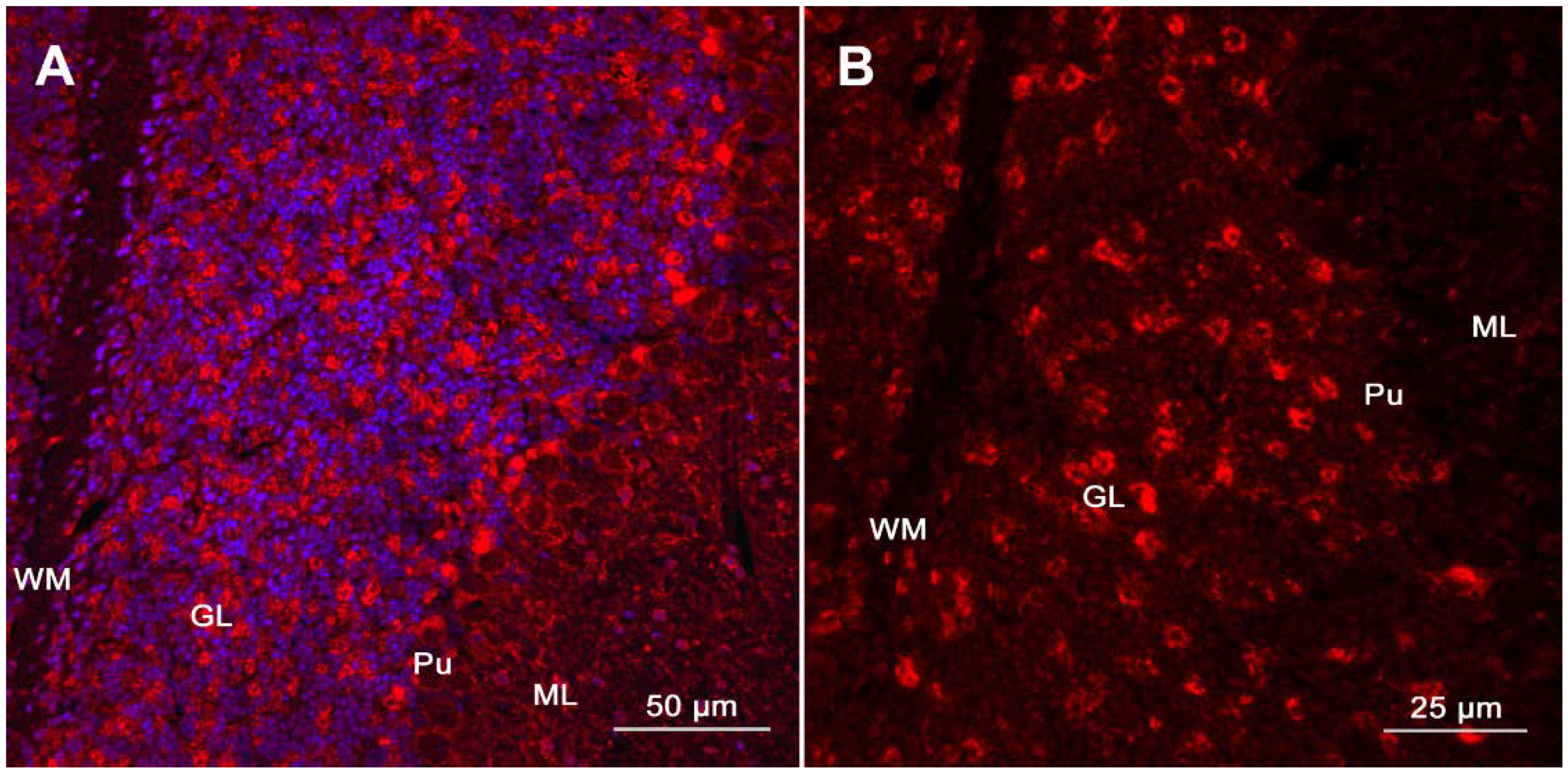{kind=link}
| Antibody Target | Neurologic Phenotype | Gender Predominance, Age-Range | Associated Cancer | Frequency of Cancer | Source |
|---|---|---|---|---|---|
| Intracellular target | |||||
| CV2/CRMP5 | EM, SNN, CA | No gender predominance, Age: 60–70 | SCLC, thymoma | >80% | [46,47,48] |
| Hu (ANNA-1) | SNN, CA, EM, LE, chronic gastrointestinal pseudo-obstruction | Slight female predominance, Age: 60–70 | SCLC > NSCLC, neuroendocrine tumors, neuroblastoma | 85% | [22,49] |
| Ma2 | LE, diencephalitis, CA, brainstem encephalitis | 70% male, Age: 60–70 in women, 30–40 in men | Testicular cancer (young men), lung cancer (older patients) | >75% | [50,51] |
| Ri (ANNA-2) | CA, OMS | Female predominance, Age: 60–70 | Breast (women) > lung cancer (men) | >70% | [22,42,52] |
| Yo (PCA-1) | CA | Almost all female, Age: 60–70 | Ovary and breast cancer | >90% | [22,40] |
| Extracellular target | |||||
| TR (DNER) | CA | >70% men, Age: 60–70 | Hodgkin lymphoma | 90% | [39,53] |
| Antibody Target | Neurologic Phenotype | Gender Predominance, Age-Range | Associated Cancer | Frequency of Cancer | Source |
|---|---|---|---|---|---|
| Intracellular target | |||||
| Amphiphysin | Polyradiculo-neuropathy, SNN, EM, SPS, CA | Slight female predominance, Age: 60–70 | SCLC, breast cancer | 80% | [54,55] |
| GAD65 | LE, SPS, CA | 70% women, Age: 50–60 | SCLC, neuroendocrine tumors, thymoma | <15% | [56,57] |
| KLHL11 | CA, brainstem syndrome | 100% men, Age: 40–50 | Testicular cancer | 80% | [58,59,60] |
| MAG n = 5 | Neuropathy, CA | 100% men, Age: 60–80 | Unknown, MGUS association | Unknown | [61] |
| PCA2 (MAP1B) | Limbic/brainstem encephalitis, LEMS, SIADH, Neuropathy, CA (37% of reported cases) | Female predominance, Age: 22–89 | SCLC, NSCLC, breast, renal, skin squamous cell, pancreas, extrapulmonary small-cell, prostate, intrahepatic primary ductal, nasopharyngeal, Ewing sarcoma, lymphoma | 80% | [62,63] |
| Extracellular target | |||||
| CASPR2 | LE, Isaac syndrome, Morvan syndrome | >70% men, Age: 60–70 | Thymoma | <30% | [64,65,66] |
| DPPX | Encephalitis, CNS hyperexcitability, PERM | >60% men, Age: 50–60 | B-cell malignancies | <10% | [67,68] |
| LGI1 | LE, CA | >60% men, Age: 60–70 | Malignant thymoma, neuroendocrine tumors | <10% | [22,69,70] |
| mGluR1 | CA, dysgeusia | No gender predominance, Age: 50–60 | Hematologic | 20–30% | [22,71] |
| P/Q VGCC | LEMS, CA | Slight female predominance, Age: 50–60 | SCLC | 50% (LEMS), 90% (in pts. w/CA) | [11,72] |
| Antibody Target | Neurologic Phenotype | Gender Predominance, Age-Range | Associated Cancer | Frequency of Cancer | Source |
|---|---|---|---|---|---|
| ARHGAP 26 (GRAF1-IgG, Anti-ca) n = 24 | subacute CA, neuropathy, psychotic symptoms, cognitive dysfunction, hyperekplexia, parkinsonism | No gender predominance, Age: 14–76 | Ovarian, Breast, Melanoma, B cell lymphoma, prostate, gastric, squamosa cell of nasopharyngeal/respiratory tract | 30–40% | [22,73,74,75] |
| CARP VIII n = 3 | CA, headache | Female predominance, Age: 69–77 | Ovarian cancer, melanoma, breast | 3/3 | [76,77,78] |
| Glycin R n = 187 | PERM/SPS 40–50%; epilepsy 20–30%; CA, movement disorders, encephalitis (30%) | No gender predominance, Age: 40–60 | Thymoma, breast cancer, Hodgkin lymphoma, SCLC, marginal B-cell lymphoma | 10–20% | [22,79] |
| Homer-3 n = 5 | CA, encephalitis, papilledema | No gender predominance, Age: 38–65 | SCLC | 1/5 | [73,80,81,82,83] |
| mGluR2 n = 2 | CA | Female predominance, Age: 3–78 | Small cell tumor, alveolar rhabdo-myosarcoma | 2/2 | [84] |
| Nb/AP3B2 n = 13 | CA, peripheral neuropathy, myelopathy | Female predominance, Age: 24–58 | Renal cell cancer, B-cell lymphoma | 2/13 | [73,85,86,87] |
| Neurochondrin n = 14 | CA, brainstem, myelopathy, psychosis, SFN | Male predominance, Age: 2–69 | Uterine cancer | 1/14 | [73,88,89,90] |
| NIF n = 41 (11 CA) | Encephalopathy, CA (27%), myelopathy, neuropathy | Male predominance, Age: 43–88 | Merkel cell carcinoma, SCLC, neuroendocrine (pancreas), Hodgkin lymphoma, hepatocellular carcinoma | 8/11 | [91] |
| PKCy n = 10 | CA | Male predominance, Age: 47–73 | NSCLC, adenocarcinoma of hepatobiliary origin | unknown | [92,93] |
| Septin-5 n = 6 | CA, oscillopsia | No gender predominance, Age: 47–72 | No association | none | [73,94] |
| SEZ6L2 n = 6 | CA, extrapyramidal symptoms, retinopathy | No gender predominance, Age: 54–69 | Breast cancer | 1/6 (4 year after CA) | [95,96,97] |
| Sj/ITPR-1 n = 23 (11 CA) | CA, polyneuropathy, encephalopathy, myelopathy | No gender predominance, Age: 7–83 | Breast, lung, renal, endometrial cancer, myeloma | 7/23 1 breast cancer 11 years after CA | [73,80,98,99,100,101] |
| SOX1 (AGNA1) n ≈ 520 (20 PCD) | LEMS (30%), CA (18.2%), limbic encephalitis (18.2%), neuropathy | Male predominance, Age: 17–87 | SCLC >> NSCLC>, Hodgkin lymphoma, breast, prostate, thyroid, esophageal cancer | >90% | [102] |
| TRIM 9, 67 n = 3 | CA, gaze palsy | No gender predominance, Age: 65–78 | Lung cancer, Melanoma | 2/2 | [103,104,105] |
| TRIM 46 n= 3 | Progressive encephalomyelitis, CA, rapidly progressive dementia | No gender predominance | SCLC | 2/3 | [104] |
| ZIC4 n = 20 | CA, OMS, SSN, dementia, SPS, brainstem encephalitis, pain, limbic encephalitis, LEMS | Male predominance | SCLC, B-cell lymphoma, multiple myeloma, breast, ovarian cancer, head and neck squamosa cell carcinoma | 14/20 | [7,106,107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loehrer, P.A.; Zieger, L.; Simon, O.J. Update on Paraneoplastic Cerebellar Degeneration. Brain Sci. 2021, 11, 1414. https://doi.org/10.3390/brainsci11111414
Loehrer PA, Zieger L, Simon OJ. Update on Paraneoplastic Cerebellar Degeneration. Brain Sciences. 2021; 11(11):1414. https://doi.org/10.3390/brainsci11111414
Chicago/Turabian StyleLoehrer, Philipp Alexander, Lara Zieger, and Ole J. Simon. 2021. "Update on Paraneoplastic Cerebellar Degeneration" Brain Sciences 11, no. 11: 1414. https://doi.org/10.3390/brainsci11111414
APA StyleLoehrer, P. A., Zieger, L., & Simon, O. J. (2021). Update on Paraneoplastic Cerebellar Degeneration. Brain Sciences, 11(11), 1414. https://doi.org/10.3390/brainsci11111414