
The Role of Mesostriatal Dopamine System and Corticostriatal Glutamatergic Transmission in Chronic Pain


{kind=link}
Abstract
1. Introduction
2. Models and Behavioral Testing of Pain in Animals
3. The Mesostriatal Dopamine System. Brain Imaging and Inactivation Experiments Implying Its Involvement in Pain Processing
4. The Effect of Nociceptive Stimuli on Dopaminergic Neurons
5. The Effect of Dopamine Deficiency on Nociception and Pain
6. The Role of Striatal Dopamine Receptors in Pain Regulation
7. Chronic Pain as a Hypodopaminergic State
8. Striatal Projection Neurons and the Effect of Dopamine on the Excitatory Inputs to the Striatum
9. Involvement of Medial Prefrontal Cortex in Pain Processing
10. Interactions of the Dopamine System with mPFC Projections to the NAc in Pain Regulation
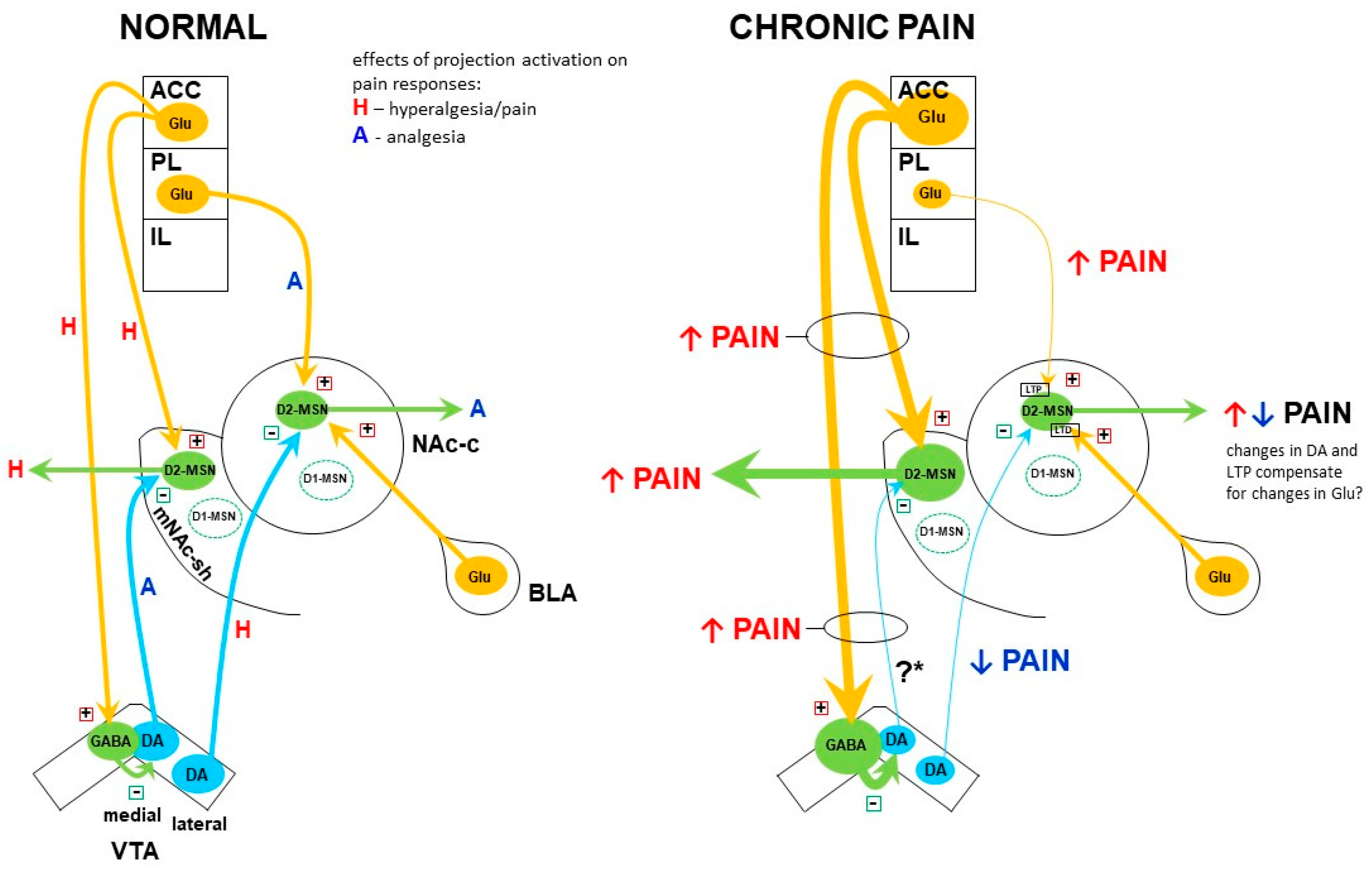
10.1. Medial NAc Shell
10.2. NAc Core
10.3. Contrasting Effects of Dopamine in the Medial NAc Shell vs. NAc Core in Pain Modulation
10.4. Alterations in the Dopamine System and Their Role in Chronic Pain—A Reappraisal
11. Altered Modulation of Dopamine Release in Chronic Pain
12. Summary
13. Methodological Issues
- (1)
- There are large differences in the duration of pain in “chronic” pain animal models. In most cases, pain duration does not exceed two weeks, and in many models, the pain lasts only a few days, whereas in humans, pain is defined as chronic when it lasts for longer than three months (or even six months) [121,122]. This raises the possibility that various experimental paradigms are not equivalent in reflecting particular stages of chronic pain development and maintenance. Chang and colleagues [22] demonstrated directly that alterations in the ventral mesostriatal system develop gradually as the pain continues and differ markedly between animals subjected to pain for five days vs. four weeks. Dias et al. [60], in turn, showed that during the transition from acute/tonic to chronic pain, there is a shift from antinociceptive to pronociceptive effect in the accumbal dopamine action on pain. On the other hand, the work by Vergara et al. [123] suggested that the ventral mesostriatal dopamine is involved in the mechanism of pain chronification but not in the maintenance of already established chronic pain. Whereas the results by Dias et al. [60] and Vergara et al. [123] are not consistent with other reports ([124]; cf also Section 5 and Section 6), perhaps due to the use of a peculiar model, they do draw attention to the fact that the modulatory effect of dopamine on pain may change as pain progresses. However, in many other studies, measurements were performed only at a single time-point and thus provide just a snapshot of dynamic changes, which may also evolve at different paces depending on the model. The changes in the mesostriatal dopaminergic activity and the dopamine–glutamate interactions described in Section 10 of this review are based on findings in models of neuropathic pain (due to sciatic nerve lesion) of five to fourteen days duration. In order to determine whether they represent the subacute, transitory, or chronic pain stage, it would be worthwhile to repeat some key experiments at much more delayed time-points, e.g., two-three months.
- (2)
- Different species (rats and mice) and strains thereof are used in the studies. While that is dictated mostly by methodological reasons (e.g., availability of transgenic mouse strains), biological differences between these organisms may be a cause of some discrepancies in pain studies. The more detailed the studies (e.g., getting down to particular subsets of projection fibers between small brain subregions), the more impact the species/strain factor is likely to have on the outcomes. Morphological differences in the dopamine system were demonstrated between inbred strains of mice, including distinct dopaminergic fibers distribution within the NAc shell [125]; even more dissimilarities in brain connectivity might be expected between mice and rats, not to mention humans.
- (3)
- The studies focus on increasingly small subregions of brain structures. When comparing data from different laboratories, it is sometimes difficult to decide if the same or distinct subregions were analyzed. For example, Ren and colleagues [63] studied dopamine effects on pain in the medial NAc shell, whereas the study by Massaly et al. [116] focused on the ventromedial NAc shell. The first study found effects on allodynia, whereas the second one excluded effect on the sensory dimension of pain but found a role of a corresponding manipulation on pain affect. It is not entirely clear if the results were dissimilar because different parts of the NAc shell were studied or because of other factors.
- (4)
- Different gene promoters are used in transgenic animals to target neuronal populations that we tend to think of as one population. In the striatum/NAc, there is the basic distinction between the two major populations of projection neurons, the indirect pathway- and the direct pathway-MSN. The D2R, adenosine receptor A2A, and proenkephalin are typical markers of the indirect pathway MSN, and promoters of these three genes are treated somewhat interchangeably to target the “D2-MSN”. However, as mentioned in Section 10.1, there is no 100% co-expression between the D2R, A2A receptor, and proenkephalin; thus, experimental manipulations on each cell population may yield different results (e.g., [126]). In the indirect pathway MSN, the D1R and prodynorphin are the analogous set of markers.
14. Concluding Remarks
Funding
Conflicts of Interest
References
- Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of Chronic Pain in Europe: Prevalence, Impact on Daily Life, and Treatment. Eur. J. Pain 2006, 10, 287–333. [Google Scholar] [CrossRef]
- Dahlhamer, J.; Lucas, J.; Zelaya, C.; Nahin, R.; Mackey, S.; DeBar, L.; Kerns, R.; Von Korff, M.; Porter, L.; Helmick, C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults—United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Nahin, R.L. Estimates of Pain Prevalence and Severity in Adults: United States, 2012. J. Pain 2015, 16, 769–780. [Google Scholar] [CrossRef] [PubMed]
- INCB. International Narcotics Control Board Report of the International Narcotics Control Board for 2019; International Narcotics Control Board: Vienna, Austria, 2019. [Google Scholar]
- O’Brien, T.; Breivik, H. The Impact of Chronic Pain-European Patients’ Perspective over 12 Months. Scand. J. Pain 2012, 3, 23–29. [Google Scholar] [CrossRef]
- Dickenson, A.H.; Suzuki, R. Opioids in Neuropathic Pain: Clues from Animal Studies. Eur. J. Pain 2005, 9, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Turk, D.C. Combining Somatic and Psychosocial Treatment for Chronic Pain Patients: Perhaps 1 + 1 Does = 3. Clin. J. Pain 2001, 17, 281–283. [Google Scholar] [CrossRef]
- IASP. International Association for the Study of Pain Terminology 2011; IASP: Washington, DC, USA, 2011. [Google Scholar]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Kuner, R. Central Mechanisms of Pathological Pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef]
- Mika, J.; Zychowska, M.; Popiolek-Barczyk, K.; Rojewska, E.; Przewlocka, B. Importance of Glial Activation in Neuropathic Pain. Eur. J. Pharmacol. 2013, 716, 106–119. [Google Scholar] [CrossRef]
- Baliki, M.N.; Apkarian, A.V. Nociception, Pain, Negative Moods, and Behavior Selection. Neuron 2015, 87, 474–491. [Google Scholar] [CrossRef]
- Burma, N.E.; Leduc-Pessah, H.; Fan, C.Y.; Trang, T. Animal Models of Chronic Pain: Advances and Challenges for Clinical Translation. J. Neurosci. Res. 2017, 95, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Coderre, T.J.; Laferrière, A. The Emergence of Animal Models of Chronic Pain and Logistical and Methodological Issues Concerning Their Use. J. Neural. Transm. 2020, 127, 393–406. [Google Scholar] [CrossRef]
- Porreca, F.; Navratilova, E. Reward, Motivation, and Emotion of Pain and Its Relief. Pain 2017, 158 (Suppl. 1), S43–S49. [Google Scholar] [CrossRef]
- Valek, L.; Auburger, G.; Tegeder, I. Sensory Neuropathy and Nociception in Rodent Models of Parkinson’s Disease. Dis. Model. Mech. 2019, 12, dmm039396. [Google Scholar] [CrossRef]
- Thompson, J.M.; Neugebauer, V. Cortico-Limbic Pain Mechanisms. Neurosci. Lett. 2019, 702, 15–23. [Google Scholar] [CrossRef]
- Ikemoto, S. Dopamine Reward Circuitry: Two Projection Systems from the Ventral Midbrain to the Nucleus Accumbens-Olfactory Tubercle Complex. Brain Res. Rev. 2007, 56, 27–78. [Google Scholar] [CrossRef]
- Lammel, S.; Lim, B.K.; Malenka, R.C. Reward and Aversion in a Heterogeneous Midbrain Dopamine System. Neuropharmacology 2014, 76 Pt B, 351–359. [Google Scholar] [CrossRef]
- Sesack, S.R.; Grace, A.A. Cortico-Basal Ganglia Reward Network: Microcircuitry. Neuropsychopharmacology 2010, 35, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Starr, C.J.; Sawaki, L.; Wittenberg, G.F.; Burdette, J.H.; Oshiro, Y.; Quevedo, A.S.; McHaffie, J.G.; Coghill, R.C. The Contribution of the Putamen to Sensory Aspects of Pain: Insights from Structural Connectivity and Brain Lesions. Brain 2011, 134, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-C.; Pollema-Mays, S.L.; Centeno, M.V.; Procissi, D.; Contini, M.; Baria, A.T.; Martina, M.; Apkarian, A.V. Role of Nucleus Accumbens in Neuropathic Pain: Linked Multi-Scale Evidence in the Rat Transitioning to Neuropathic Pain. Pain 2014, 155, 1128–1139. [Google Scholar] [CrossRef]
- Magnusson, J.E.; Martin, R.V. Additional Evidence for the Involvement of the Basal Ganglia in Formalin-Induced Nociception: The Role of the Nucleus Accumbens. Brain Res. 2002, 942, 128–132. [Google Scholar] [CrossRef]
- Baliki, M.N.; Petre, B.; Torbey, S.; Herrmann, K.M.; Huang, L.; Schnitzer, T.J.; Fields, H.L.; Apkarian, A.V. Corticostriatal Functional Connectivity Predicts Transition to Chronic Back Pain. Nat. Neurosci. 2012, 15, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Geha, P.Y.; Baliki, M.N.; Harden, R.N.; Bauer, W.R.; Parrish, T.B.; Apkarian, A.V. The Brain in Chronic CRPS Pain: Abnormal Gray-White Matter Interactions in Emotional and Autonomic Regions. Neuron 2008, 60, 570–581. [Google Scholar] [CrossRef]
- Makary, M.M.; Polosecki, P.; Cecchi, G.A.; DeAraujo, I.E.; Barron, D.S.; Constable, T.R.; Whang, P.G.; Thomas, D.A.; Mowafi, H.; Small, D.M.; et al. Loss of Nucleus Accumbens Low-Frequency Fluctuations Is a Signature of Chronic Pain. Proc. Natl. Acad. Sci. USA 2020, 117, 10015–10023. [Google Scholar] [CrossRef]
- Baliki, M.N.; Geha, P.Y.; Fields, H.L.; Apkarian, A.V. Predicting Value of Pain and Analgesia: Nucleus Accumbens Response to Noxious Stimuli Changes in the Presence of Chronic Pain. Neuron 2010, 66, 149–160. [Google Scholar] [CrossRef]
- Ungless, M.A.; Magill, P.J.; Bolam, J.P. Uniform Inhibition of Dopamine Neurons in the Ventral Tegmental Area by Aversive Stimuli. Science 2004, 303, 2040–2042. [Google Scholar] [CrossRef]
- Brischoux, F.; Chakraborty, S.; Brierley, D.I.; Ungless, M.A. Phasic Excitation of Dopamine Neurons in Ventral VTA by Noxious Stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 4894–4899. [Google Scholar] [CrossRef]
- Budygin, E.A.; Park, J.; Bass, C.E.; Grinevich, V.P.; Bonin, K.D.; Wightman, R.M. Aversive Stimulus Differentially Triggers Subsecond Dopamine Release in Reward Regions. Neuroscience 2012, 201, 331–337. [Google Scholar] [CrossRef]
- Park, J.; Bucher, E.S.; Budygin, E.A.; Wightman, R.M. Norepinephrine and Dopamine Transmission in 2 Limbic Regions Differentially Respond to Acute Noxious Stimulation. Pain 2015, 156, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.J.; Heitzeg, M.M.; Koeppe, R.A.; Stohler, C.S.; Zubieta, J.-K. Variations in the Human Pain Stress Experience Mediated by Ventral and Dorsal Basal Ganglia Dopamine Activity. J. Neurosci. 2006, 26, 10789–10795. [Google Scholar] [CrossRef]
- Wood, P.B.; Schweinhardt, P.; Jaeger, E.; Dagher, A.; Hakyemez, H.; Rabiner, E.A.; Bushnell, M.C.; Chizh, B.A. Fibromyalgia Patients Show an Abnormal Dopamine Response to Pain. Eur. J. Neurosci. 2007, 25, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Beiske, A.G.; Loge, J.H.; Rønningen, A.; Svensson, E. Pain in Parkinson’s Disease: Prevalence and Characteristics. Pain 2009, 141, 173–177. [Google Scholar] [CrossRef]
- Ford, B. Pain in Parkinson’s Disease. Mov. Disord. 2010, 25 (Suppl. 1), S98–S103. [Google Scholar] [CrossRef]
- Sung, S.; Vijiaratnam, N.; Chan, D.W.C.; Farrell, M.; Evans, A.H. Pain Sensitivity in Parkinson’s Disease: Systematic Review and Meta-Analysis. Parkinsonism Relat. Disord. 2018, 48, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Zambito Marsala, S.; Tinazzi, M.; Vitaliani, R.; Recchia, S.; Fabris, F.; Marchini, C.; Fiaschi, A.; Moretto, G.; Giometto, B.; Macerollo, A.; et al. Spontaneous Pain, Pain Threshold, and Pain Tolerance in Parkinson’s Disease. J. Neurol. 2011, 258, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Vijiaratnam, N.; Chan, D.W.C.; Farrell, M.; Evans, A.H. Parkinson Disease: A Systemic Review of Pain Sensitivities and Its Association with Clinical Pain and Response to Dopaminergic Stimulation. J. Neurol. Sci. 2018, 395, 172–206. [Google Scholar] [CrossRef]
- Chudler, E.H.; Lu, Y. Nociceptive Behavioral Responses to Chemical, Thermal and Mechanical Stimulation after Unilateral, Intrastriatal Administration of 6-Hydroxydopamine. Brain Res. 2008, 1213, 41–47. [Google Scholar] [CrossRef]
- Gee, L.E.; Chen, N.; Ramirez-Zamora, A.; Shin, D.S.; Pilitsis, J.G. The Effects of Subthalamic Deep Brain Stimulation on Mechanical and Thermal Thresholds in 6OHDA-Lesioned Rats. Eur. J. Neurosci. 2015, 42, 2061–2069. [Google Scholar] [CrossRef]
- Lin, M.T.; Wu, J.J.; Chandra, A.; Tsay, B.L. Activation of Striatal Dopamine Receptors Induces Pain Inhibition in Rats. J. Neural. Transm. 1981, 51, 213–222. [Google Scholar] [CrossRef]
- Saadé, N.E.; Atweh, S.F.; Bahuth, N.B.; Jabbur, S.J. Augmentation of Nociceptive Reflexes and Chronic Deafferentation Pain by Chemical Lesions of Either Dopaminergic Terminals or Midbrain Dopaminergic Neurons. Brain Res. 1997, 751, 1–12. [Google Scholar] [CrossRef]
- Takeda, R.; Ikeda, T.; Tsuda, F.; Abe, H.; Hashiguchi, H.; Ishida, Y.; Nishimori, T. Unilateral Lesions of Mesostriatal Dopaminergic Pathway Alters the Withdrawal Response of the Rat Hindpaw to Mechanical Stimulation. Neurosci. Res. 2005, 52, 31–36. [Google Scholar] [CrossRef]
- Dieb, W.; Ouachikh, O.; Alves, S.; Boucher, Y.; Durif, F.; Hafidi, A. Nigrostriatal Dopaminergic Depletion Increases Static Orofacial Allodynia. J. Headache Pain 2016, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Dieb, W.; Ouachikh, O.; Durif, F.; Hafidi, A. Lesion of the Dopaminergic Nigrostriatal Pathway Induces Trigeminal Dynamic Mechanical Allodynia. Brain Behav. 2014, 4, 368–380. [Google Scholar] [CrossRef]
- Park, J.; Lim, C.-S.; Seo, H.; Park, C.-A.; Zhuo, M.; Kaang, B.-K.; Lee, K. Pain Perception in Acute Model Mice of Parkinson’s Disease Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP). Mol. Pain 2015, 11, 28. [Google Scholar] [CrossRef]
- Rosland, J.H.; Hunskaar, S.; Broch, O.J.; Hole, K. Acute and Long Term Effects of 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) in Tests of Nociception in Mice. Pharmacol. Toxicol. 1992, 70, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Franklin, K.B. 6-Hydroxydopamine Lesions of the Ventral Tegmentum Abolish D-Amphetamine and Morphine Analgesia in the Formalin Test but Not in the Tail Flick Test. Brain Res. 1990, 519, 144–149. [Google Scholar] [CrossRef]
- Jurna, I.; Heinz, G.; Blinn, G.; Nell, T. The Effect of Substantia Negra Stimulation and Morphine on Alpha-Motoneurones and the Tail-Flick Response. Eur. J. Pharmacol. 1978, 51, 239–250. [Google Scholar] [CrossRef]
- Li, A.-L.; Sibi, J.E.; Yang, X.; Chiao, J.-C.; Peng, Y.B. Stimulation of the Ventral Tegmental Area Increased Nociceptive Thresholds and Decreased Spinal Dorsal Horn Neuronal Activity in Rat. Exp. Brain Res. 2016, 234, 1505–1514. [Google Scholar] [CrossRef]
- Sandberg, D.E.; Segal, M. Pharmacological Analysis of Analgesia and Self-Stimulation Elicited by Electrical Stimulation of Catecholamine Nuclei in the Rat Brain. Brain Res. 1978, 152, 529–542. [Google Scholar] [CrossRef]
- Taylor, N.E.; Long, H.; Pei, J.; Kukutla, P.; Phero, A.; Hadaegh, F.; Abdelnabi, A.; Solt, K.; Brenner, G.J. The Rostromedial Tegmental Nucleus: A Key Modulator of Pain and Opioid Analgesia. Pain 2019, 160, 2524–2534. [Google Scholar] [CrossRef]
- Watanabe, M.; Narita, M.; Hamada, Y.; Yamashita, A.; Tamura, H.; Ikegami, D.; Kondo, T.; Shinzato, T.; Shimizu, T.; Fukuchi, Y.; et al. Activation of Ventral Tegmental Area Dopaminergic Neurons Reverses Pathological Allodynia Resulting from Nerve Injury or Bone Cancer. Mol. Pain 2018, 14, 1744806918756406. [Google Scholar] [CrossRef] [PubMed]
- Barrot, M.; Sesack, S.R.; Georges, F.; Pistis, M.; Hong, S.; Jhou, T.C. Braking Dopamine Systems: A New GABA Master Structure for Mesolimbic and Nigrostriatal Functions. J. Neurosci. 2012, 32, 14094–14101. [Google Scholar] [CrossRef] [PubMed]
- Bourdy, R.; Barrot, M. A New Control Center for Dopaminergic Systems: Pulling the VTA by the Tail. Trends Neurosci. 2012, 35, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Jhou, T.C. The Rostromedial Tegmental (RMTg) “Brake” on Dopamine and Behavior: A Decade of Progress but Also Much Unfinished Work. Neuropharmacology 2021, 198, 108763. [Google Scholar] [CrossRef] [PubMed]
- Jhou, T.C.; Geisler, S.; Marinelli, M.; Degarmo, B.A.; Zahm, D.S. The Mesopontine Rostromedial Tegmental Nucleus: A Structure Targeted by the Lateral Habenula That Projects to the Ventral Tegmental Area of Tsai and Substantia Nigra Compacta. J. Comp. Neurol. 2009, 513, 566–596. [Google Scholar] [CrossRef] [PubMed]
- Altier, N.; Stewart, J. The Role of Dopamine in the Nucleus Accumbens in Analgesia. Life Sci. 1999, 65, 2269–2287. [Google Scholar] [CrossRef]
- Ansah, O.B.; Leite-Almeida, H.; Wei, H.; Pertovaara, A. Striatal Dopamine D2 Receptors Attenuate Neuropathic Hypersensitivity in the Rat. Exp. Neurol. 2007, 205, 536–546. [Google Scholar] [CrossRef]
- Dias, E.V.; Sartori, C.R.; Marião, P.R.; Vieira, A.S.; Camargo, L.C.; Athie, M.C.P.; Pagliusi, M.O.; Tambeli, C.H.; Parada, C.A. Nucleus Accumbens Dopaminergic Neurotransmission Switches Its Modulatory Action in Chronification of Inflammatory Hyperalgesia. Eur. J. Neurosci. 2015, 42, 2380–2389. [Google Scholar] [CrossRef]
- Magnusson, J.E.; Fisher, K. The Involvement of Dopamine in Nociception: The Role of D(1) and D(2) Receptors in the Dorsolateral Striatum. Brain Res. 2000, 855, 260–266. [Google Scholar] [CrossRef]
- Mlost, J.; Wąsik, A.; Michaluk, J.T.; Antkiewicz-Michaluk, L.; Starowicz, K. Changes in Monoaminergic Neurotransmission in an Animal Model of Osteoarthritis: The Role of Endocannabinoid Signaling. Front. Mol. Neurosci. 2018, 11, 466. [Google Scholar] [CrossRef]
- Ren, W.; Centeno, M.V.; Berger, S.; Wu, Y.; Na, X.; Liu, X.; Kondapalli, J.; Apkarian, A.V.; Martina, M.; Surmeier, D.J. The Indirect Pathway of the Nucleus Accumbens Shell Amplifies Neuropathic Pain. Nat. Neurosci. 2016, 19, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.W.; Murphy, N.P.; Evans, C.J.; Cahill, C.M. Correlation between Ventral Striatal Catecholamine Content and Nociceptive Thresholds in Neuropathic Mice. J. Pain 2014, 15, 878–885. [Google Scholar] [CrossRef]
- Wu, Y.; Na, X.; Zang, Y.; Cui, Y.; Xin, W.; Pang, R.; Zhou, L.; Wei, X.; Li, Y.; Liu, X. Upregulation of Tumor Necrosis Factor-Alpha in Nucleus Accumbens Attenuates Morphine-Induced Rewarding in a Neuropathic Pain Model. Biochem. Biophys. Res. Commun. 2014, 449, 502–507. [Google Scholar] [CrossRef]
- Huang, S.; Borgland, S.L.; Zamponi, G.W. Peripheral Nerve Injury-Induced Alterations in VTA Neuron Firing Properties. Mol. Brain 2019, 12, 89. [Google Scholar] [CrossRef]
- Ren, W.; Centeno, M.V.; Wei, X.; Wickersham, I.; Martina, M.; Apkarian, A.V.; Surmeier, D.J. Adaptive Alterations in the Mesoaccumbal Network after Peripheral Nerve Injury. Pain 2021, 162, 895–906. [Google Scholar] [CrossRef]
- Martikainen, I.K.; Nuechterlein, E.B.; Peciña, M.; Love, T.M.; Cummiford, C.M.; Green, C.R.; Stohler, C.S.; Zubieta, J.-K. Chronic Back Pain Is Associated with Alterations in Dopamine Neurotransmission in the Ventral Striatum. J. Neurosci. 2015, 35, 9957–9965. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Narita, M.; Narita, M.; Iino, M.; Sugita, J.; Matsumura, Y.; Suzuki, T. Suppression of the Morphine-Induced Rewarding Effect in the Rat with Neuropathic Pain: Implication of the Reduction in Mu-Opioid Receptor Functions in the Ventral Tegmental Area. J. Neurochem. 2002, 82, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kishimoto, Y.; Misawa, M. Formalin- and Carrageenan-Induced Inflammation Attenuates Place Preferences Produced by Morphine, Methamphetamine and Cocaine. Life Sci. 1996, 59, 1667–1674. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J.; Sibley, D.R. D1 and D2 Dopamine Receptor-Regulated Gene Expression of Striatonigral and Striatopallidal Neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B. The Functional Anatomy of Basal Ganglia Disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Surmeier, D.J. Modulation of Striatal Projection Systems by Dopamine. Ann. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef] [PubMed]
- Kupchik, Y.M.; Brown, R.M.; Heinsbroek, J.A.; Lobo, M.K.; Schwartz, D.J.; Kalivas, P.W. Coding the Direct/Indirect Pathways by D1 and D2 Receptors Is Not Valid for Accumbens Projections. Nat. Neurosci. 2015, 18, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Furuta, T.; Kaneko, T. Chemical Organization of Projection Neurons in the Rat Accumbens Nucleus and Olfactory Tubercle. Neuroscience 2003, 120, 783–798. [Google Scholar] [CrossRef]
- Curran, E.J.; Watson, S.J. Dopamine Receptor MRNA Expression Patterns by Opioid Peptide Cells in the Nucleus Accumbens of the Rat: A Double in Situ Hybridization Study. J. Comp. Neurol. 1995, 361, 57–76. [Google Scholar] [CrossRef]
- Perreault, M.L.; Hasbi, A.; Alijaniaram, M.; Fan, T.; Varghese, G.; Fletcher, P.J.; Seeman, P.; O’Dowd, B.F.; George, S.R. The Dopamine D1-D2 Receptor Heteromer Localizes in Dynorphin/Enkephalin Neurons: Increased High Affinity State Following Amphetamine and in Schizophrenia. J. Biol. Chem. 2010, 285, 36625–36634. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 Dopamine-Receptor Modulation of Striatal Glutamatergic Signaling in Striatal Medium Spiny Neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef]
- Yin, H.H.; Lovinger, D.M. Frequency-Specific and D2 Receptor-Mediated Inhibition of Glutamate Release by Retrograde Endocannabinoid Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 8251–8256. [Google Scholar] [CrossRef] [PubMed]
- Baliki, M.N.; Chialvo, D.R.; Geha, P.Y.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Apkarian, A.V. Chronic Pain and the Emotional Brain: Specific Brain Activity Associated with Spontaneous Fluctuations of Intensity of Chronic Back Pain. J. Neurosci. 2006, 26, 12165–12173. [Google Scholar] [CrossRef]
- Lee, M.; Manders, T.R.; Eberle, S.E.; Su, C.; D’amour, J.; Yang, R.; Lin, H.Y.; Deisseroth, K.; Froemke, R.C.; Wang, J. Activation of Corticostriatal Circuitry Relieves Chronic Neuropathic Pain. J. Neurosci. 2015, 35, 5247–5259. [Google Scholar] [CrossRef]
- Wang, G.-Q.; Cen, C.; Li, C.; Cao, S.; Wang, N.; Zhou, Z.; Liu, X.-M.; Xu, Y.; Tian, N.-X.; Zhang, Y.; et al. Deactivation of Excitatory Neurons in the Prelimbic Cortex via Cdk5 Promotes Pain Sensation and Anxiety. Nat. Commun. 2015, 6, 7660. [Google Scholar] [CrossRef]
- Zhang, Z.; Gadotti, V.M.; Chen, L.; Souza, I.A.; Stemkowski, P.L.; Zamponi, G.W. Role of Prelimbic GABAergic Circuits in Sensory and Emotional Aspects of Neuropathic Pain. Cell Rep. 2015, 12, 752–759. [Google Scholar] [CrossRef]
- Chen, T.; Taniguchi, W.; Chen, Q.-Y.; Tozaki-Saitoh, H.; Song, Q.; Liu, R.-H.; Koga, K.; Matsuda, T.; Kaito-Sugimura, Y.; Wang, J.; et al. Top-down Descending Facilitation of Spinal Sensory Excitatory Transmission from the Anterior Cingulate Cortex. Nat. Commun. 2018, 9, 1886. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-H.; Shen, L.-L.; Wen, H.-Z.; Zhao, Y.-D.; Chen, P.-H.; Ruan, H.-Z. The Projections from the Anterior Cingulate Cortex to the Nucleus Accumbens and Ventral Tegmental Area Contribute to Neuropathic Pain-Evoked Aversion in Rats. Neurobiol. Dis. 2020, 140, 104862. [Google Scholar] [CrossRef]
- Gu, L.; Uhelski, M.L.; Anand, S.; Romero-Ortega, M.; Kim, Y.; Fuchs, P.N.; Mohanty, S.K. Pain Inhibition by Optogenetic Activation of Specific Anterior Cingulate Cortical Neurons. PLoS ONE 2015, 10, e0117746. [Google Scholar] [CrossRef]
- Johansen, J.P.; Fields, H.L. Glutamatergic Activation of Anterior Cingulate Cortex Produces an Aversive Teaching Signal. Nat. Neurosci. 2004, 7, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Ko, H.-G.; Chen, T.; Descalzi, G.; Koga, K.; Wang, H.; Kim, S.S.; Shang, Y.; Kwak, C.; Park, S.-W.; et al. Alleviating Neuropathic Pain Hypersensitivity by Inhibiting PKMzeta in the Anterior Cingulate Cortex. Science 2010, 330, 1400–1404. [Google Scholar] [CrossRef]
- Schwartz, N.; Temkin, P.; Jurado, S.; Lim, B.K.; Heifets, B.D.; Polepalli, J.S.; Malenka, R.C. Chronic Pain. Decreased Motivation during Chronic Pain Requires Long-Term Depression in the Nucleus Accumbens. Science 2014, 345, 535–542. [Google Scholar] [CrossRef]
- Ji, G.; Sun, H.; Fu, Y.; Li, Z.; Pais-Vieira, M.; Galhardo, V.; Neugebauer, V. Cognitive Impairment in Pain through Amygdala-Driven Prefrontal Cortical Deactivation. J. Neurosci. 2010, 30, 5451–5464. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Huang, M.; Meltzer, H.; Martina, M. Reduced Glutamatergic Currents and Dendritic Branching of Layer 5 Pyramidal Cells Contribute to Medial Prefrontal Cortex Deactivation in a Rat Model of Neuropathic Pain. Front. Cell Neurosci. 2016, 10, 133. [Google Scholar] [CrossRef]
- Chao, T.-H.H.; Chen, J.-H.; Yen, C.-T. Plasticity Changes in Forebrain Activity and Functional Connectivity during Neuropathic Pain Development in Rats with Sciatic Spared Nerve Injury. Mol. Brain 2018, 11, 55. [Google Scholar] [CrossRef]
- Zhao, R.; Zhou, H.; Huang, L.; Xie, Z.; Wang, J.; Gan, W.-B.; Yang, G. Neuropathic Pain Causes Pyramidal Neuronal Hyperactivity in the Anterior Cingulate Cortex. Front. Cell Neurosci. 2018, 12, 107. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Q.; Martinez, E.; Dale, J.; Hu, S.; Zhang, E.; Liu, K.; Huang, D.; Yang, G.; Chen, Z.; et al. Ketamine Reduces Aversion in Rodent Pain Models by Suppressing Hyperactivity of the Anterior Cingulate Cortex. Nat. Commun. 2018, 9, 3751. [Google Scholar] [CrossRef]
- Meda, K.S.; Patel, T.; Braz, J.M.; Malik, R.; Turner, M.L.; Seifikar, H.; Basbaum, A.I.; Sohal, V.S. Microcircuit Mechanisms through Which Mediodorsal Thalamic Input to Anterior Cingulate Cortex Exacerbates Pain-Related Aversion. Neuron 2019, 102, 944–959.e3. [Google Scholar] [CrossRef] [PubMed]
- Mussio, C.A.; Harte, S.E.; Borszcz, G.S. Regional Differences Within the Anterior Cingulate Cortex in the Generation Versus Suppression of Pain Affect in Rats. J. Pain 2020, 21, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mitrić, M.; Seewald, A.; Moschetti, G.; Sacerdote, P.; Ferraguti, F.; Kummer, K.K.; Kress, M. Layer- and Subregion-Specific Electrophysiological and Morphological Changes of the Medial Prefrontal Cortex in a Mouse Model of Neuropathic Pain. Sci. Rep. 2019, 9, 9479. [Google Scholar] [CrossRef] [PubMed]
- David-Pereira, A.; Puga, S.; Gonçalves, S.; Amorim, D.; Silva, C.; Pertovaara, A.; Almeida, A.; Pinto-Ribeiro, F. Metabotropic Glutamate 5 Receptor in the Infralimbic Cortex Contributes to Descending Pain Facilitation in Healthy and Arthritic Animals. Neuroscience 2016, 312, 108–119. [Google Scholar] [CrossRef]
- Kiritoshi, T.; Ji, G.; Neugebauer, V. Rescue of Impaired MGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats. J. Neurosci. 2016, 36, 837–850. [Google Scholar] [CrossRef]
- Ji, G.; Neugebauer, V. Modulation of Medial Prefrontal Cortical Activity Using in Vivo Recordings and Optogenetics. Mol. Brain 2012, 5, 36. [Google Scholar] [CrossRef]
- Brog, J.S.; Salyapongse, A.; Deutch, A.Y.; Zahm, D.S. The Patterns of Afferent Innervation of the Core and Shell in the “Accumbens” Part of the Rat Ventral Striatum: Immunohistochemical Detection of Retrogradely Transported Fluoro-Gold. J. Comp. Neurol. 1993, 338, 255–278. [Google Scholar] [CrossRef]
- Anagnostakis, Y.; Zis, V.; Spyraki, C. Analgesia Induced by Morphine Injected into the Pallidum. Behav. Brain Res. 1992, 48, 135–143. [Google Scholar] [CrossRef]
- Kupchik, Y.M.; Scofield, M.D.; Rice, K.C.; Cheng, K.; Roques, B.P.; Kalivas, P.W. Cocaine Dysregulates Opioid Gating of GABA Neurotransmission in the Ventral Pallidum. J. Neurosci. 2014, 34, 1057–1066. [Google Scholar] [CrossRef]
- Sagheddu, C.; Aroni, S.; De Felice, M.; Lecca, S.; Luchicchi, A.; Melis, M.; Muntoni, A.L.; Romano, R.; Palazzo, E.; Guida, F.; et al. Enhanced Serotonin and Mesolimbic Dopamine Transmissions in a Rat Model of Neuropathic Pain. Neuropharmacology 2015, 97, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Opposite Effects of Mu and Kappa Opiate Agonists on Dopamine Release in the Nucleus Accumbens and in the Dorsal Caudate of Freely Moving Rats. J. Pharmacol. Exp. Ther. 1988, 244, 1067–1080. [Google Scholar]
- Ford, C.P. The Role of D2-Autoreceptors in Regulating Dopamine Neuron Activity and Transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Spanagel, R.; Herz, A.; Shippenberg, T.S. Opposing Tonically Active Endogenous Opioid Systems Modulate the Mesolimbic Dopaminergic Pathway. Proc. Natl. Acad. Sci. USA 1992, 89, 2046–2050. [Google Scholar] [CrossRef]
- Selley, D.E.; Lazenka, M.F.; Sim-Selley, L.J.; Secor McVoy, J.R.; Potter, D.N.; Chartoff, E.H.; Carlezon, W.A.; Negus, S.S. Attenuated Dopamine Receptor Signaling in Nucleus Accumbens Core in a Rat Model of Chemically-Induced Neuropathy. Neuropharmacology 2020, 166, 107935. [Google Scholar] [CrossRef] [PubMed]
- Wawrzczak-Bargieła, A.; Ziółkowska, B.; Piotrowska, A.; Starnowska-Sokół, J.; Rojewska, E.; Mika, J.; Przewłocka, B.; Przewłocki, R. Neuropathic Pain Dysregulates Gene Expression of the Forebrain Opioid and Dopamine Systems. Neurotox. Res. 2020, 37, 800–814. [Google Scholar] [CrossRef]
- Austin, P.J.; Beyer, K.; Bembrick, A.L.; Keay, K.A. Peripheral Nerve Injury Differentially Regulates Dopaminergic Pathways in the Nucleus Accumbens of Rats with Either “pain Alone” or “Pain and Disability”. Neuroscience 2010, 171, 329–343. [Google Scholar] [CrossRef]
- Bals-Kubik, R.; Herz, A.; Shippenberg, T.S. Evidence That the Aversive Effects of Opioid Antagonists and Kappa-Agonists Are Centrally Mediated. Psychopharmacology 1989, 98, 203–206. [Google Scholar] [CrossRef]
- Knoll, A.T.; Carlezon, W.A. Dynorphin, Stress, and Depression. Brain Res. 2010, 1314, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Tejeda, H.A.; Shippenberg, T.S.; Henriksson, R. The Dynorphin/κ-Opioid Receptor System and Its Role in Psychiatric Disorders. Cell Mol. Life Sci. 2012, 69, 857–896. [Google Scholar] [CrossRef] [PubMed]
- Shippenberg, T.S.; Zapata, A.; Chefer, V.I. Dynorphin and the Pathophysiology of Drug Addiction. Pharmacol. Ther. 2007, 116, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Pickens, S.; Burma, N.E.; Ibarra-Lecue, I.; Yang, H.; Xue, L.; Cook, C.; Hakimian, J.K.; Severino, A.L.; Lueptow, L.; et al. Kappa Opioid Receptors Drive a Tonic Aversive Component of Chronic Pain. J. Neurosci. 2019, 39, 4162–4178. [Google Scholar] [CrossRef]
- Massaly, N.; Copits, B.A.; Wilson-Poe, A.R.; Hipólito, L.; Markovic, T.; Yoon, H.J.; Liu, S.; Walicki, M.C.; Bhatti, D.L.; Sirohi, S.; et al. Pain-Induced Negative Affect Is Mediated via Recruitment of The Nucleus Accumbens Kappa Opioid System. Neuron 2019, 102, 564–573.e6. [Google Scholar] [CrossRef]
- Narita, M.; Kishimoto, Y.; Ise, Y.; Yajima, Y.; Misawa, K.; Suzuki, T. Direct Evidence for the Involvement of the Mesolimbic Kappa-Opioid System in the Morphine-Induced Rewarding Effect under an Inflammatory Pain-like State. Neuropsychopharmacology 2005, 30, 111–118. [Google Scholar] [CrossRef]
- Suzuki, T.; Kishimoto, Y.; Misawa, M.; Nagase, H.; Takeda, F. Role of the Kappa-Opioid System in the Attenuation of the Morphine-Induced Place Preference under Chronic Pain. Life Sci. 1999, 64, PL1–PL7. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.W.; Becker, S.; Schweinhardt, P.; Cahill, C. Mesolimbic Dopamine Signaling in Acute and Chronic Pain: Implications for Motivation, Analgesia, and Addiction. Pain 2016, 157, 1194–1198. [Google Scholar] [CrossRef]
- Watanabe, M.; Narita, M. Brain Reward Circuit and Pain. Adv. Exp. Med. Biol. 2018, 1099, 201–210. [Google Scholar] [CrossRef]
- Katz, J.; Rosenbloom, B.N.; Fashler, S. Chronic Pain, Psychopathology, and DSM-5 Somatic Symptom Disorder. Can. J. Psychiatry 2015, 60, 160–167. [Google Scholar] [CrossRef]
- Treede, R.-D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. A Classification of Chronic Pain for ICD-11. Pain 2015, 156, 1003–1007. [Google Scholar] [CrossRef]
- Vergara, F.; Sardi, N.F.; Pescador, A.C.; Guaita, G.O.; Jark Stern, C.A.; Chichorro, J.G.; Fischer, L. Contribution of Mesolimbic Dopamine and Kappa Opioid Systems to the Transition from Acute to Chronic Pain. Neuropharmacology 2020, 178, 108226. [Google Scholar] [CrossRef]
- Sarkis, R.; Saadé, N.; Atweh, S.; Jabbur, S.; Al-Amin, H. Chronic Dizocilpine or Apomorphine and Development of Neuropathy in Two Rat Models I: Behavioral Effects and Role of Nucleus Accumbens. Exp. Neurol. 2011, 228, 19–29. [Google Scholar] [CrossRef]
- D’Este, L.; Casini, A.; Puglisi-Allegra, S.; Cabib, S.; Renda, T.G. Comparative Immunohistochemical Study of the Dopaminergic Systems in Two Inbred Mouse Strains (C57BL/6J and DBA/2J). J. Chem. Neuroanat. 2007, 33, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Severino, A.L.; Mittal, N.; Hakimian, J.K.; Velarde, N.; Minasyan, A.; Albert, R.; Torres, C.; Romaneschi, N.; Johnston, C.; Tiwari, S.; et al. μ-Opioid Receptors on Distinct Neuronal Populations Mediate Different Aspects of Opioid Reward-Related Behaviors. eNeuro 2020, 7, ENEURO.0146-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Goffer, Y.; Xu, D.; Eberle, S.E.; D’amour, J.; Lee, M.; Tukey, D.; Froemke, R.C.; Ziff, E.B.; Wang, J. Calcium-Permeable AMPA Receptors in the Nucleus Accumbens Regulate Depression-like Behaviors in the Chronic Neuropathic Pain State. J. Neurosci. 2013, 33, 19034–19044. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; D’amour, J.; Lee, M.; Lin, H.-Y.; Manders, T.; Xu, D.; Eberle, S.E.; Goffer, Y.; Zou, A.H.; Rahman, M.; et al. Persistent Pain Alters AMPA Receptor Subunit Levels in the Nucleus Accumbens. Mol. Brain 2015, 8, 46. [Google Scholar] [CrossRef]
- Bruera, E.; Chadwick, S.; Brenneis, C.; Hanson, J.; MacDonald, R.N. Methylphenidate Associated with Narcotics for the Treatment of Cancer Pain. Cancer Treat. Rep. 1987, 71, 67–70. [Google Scholar]
- Forrest, W.H.; Brown, B.W.; Brown, C.R.; Defalque, R.; Gold, M.; Gordon, H.E.; James, K.E.; Katz, J.; Mahler, D.L.; Schroff, P.; et al. Dextroamphetamine with Morphine for the Treatment of Postoperative Pain. N. Engl. J. Med. 1977, 296, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.; Melzack, R. Psychostimulant Drugs Potentiate Morphine Analgesia in the Formalin Test. J. Pain Symptom Manag. 1998, 16, 230–239. [Google Scholar] [CrossRef]
- Engeln, M.; Ahmed, S.H.; Vouillac, C.; Tison, F.; Bezard, E.; Fernagut, P.-O. Reinforcing Properties of Pramipexole in Normal and Parkinsonian Rats. Neurobiol. Dis. 2013, 49, 79–86. [Google Scholar] [CrossRef]
- Holman, A.J.; Myers, R.R. A Randomized, Double-Blind, Placebo-Controlled Trial of Pramipexole, a Dopamine Agonist, in Patients with Fibromyalgia Receiving Concomitant Medications. Arthritis Rheum. 2005, 52, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Miley, D.P.; Abrams, A.A.; Atkinson, J.H.; Janowsky, D.S. Successful Treatment of Thalamic Pain with Apomorphine. Am. J. Psychiatry 1978, 135, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Stuginski-Barbosa, J.; Rodrigues, G.G.R.; Bigal, M.E.; Speciali, J.G. Burning Mouth Syndrome Responsive to Pramipexol. J. Headache Pain 2008, 9, 43–45. [Google Scholar] [CrossRef] [PubMed][Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziółkowska, B. The Role of Mesostriatal Dopamine System and Corticostriatal Glutamatergic Transmission in Chronic Pain. Brain Sci. 2021, 11, 1311. https://doi.org/10.3390/brainsci11101311
Ziółkowska B. The Role of Mesostriatal Dopamine System and Corticostriatal Glutamatergic Transmission in Chronic Pain. Brain Sciences. 2021; 11(10):1311. https://doi.org/10.3390/brainsci11101311
Chicago/Turabian StyleZiółkowska, Barbara. 2021. "The Role of Mesostriatal Dopamine System and Corticostriatal Glutamatergic Transmission in Chronic Pain" Brain Sciences 11, no. 10: 1311. https://doi.org/10.3390/brainsci11101311
APA StyleZiółkowska, B. (2021). The Role of Mesostriatal Dopamine System and Corticostriatal Glutamatergic Transmission in Chronic Pain. Brain Sciences, 11(10), 1311. https://doi.org/10.3390/brainsci11101311