Plutonium Migration during the Leaching of Cemented Radioactive Waste Sludges

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Safety
2.2. Preparation of Plutonium Solutions
2.3. Cement Preparation
2.4. Leaching Experiments
2.5. Micro-Focus X-Ray Fluorescence (XRF) and X-Ray Absorption Spectroscopy (XAS)
3. Results and Discussion
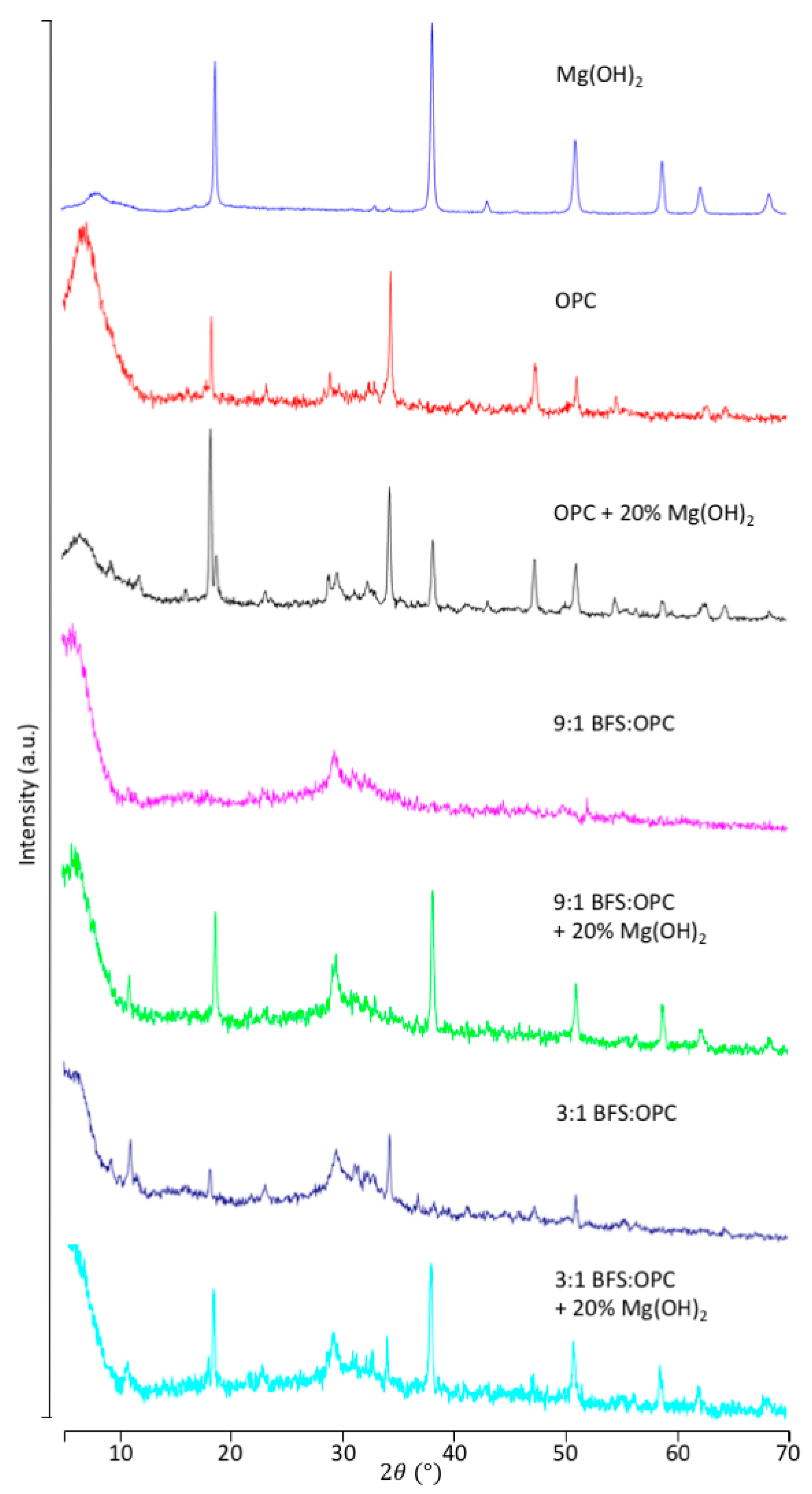
3.1. Incorporation of Mg(OH)2 in the Cement
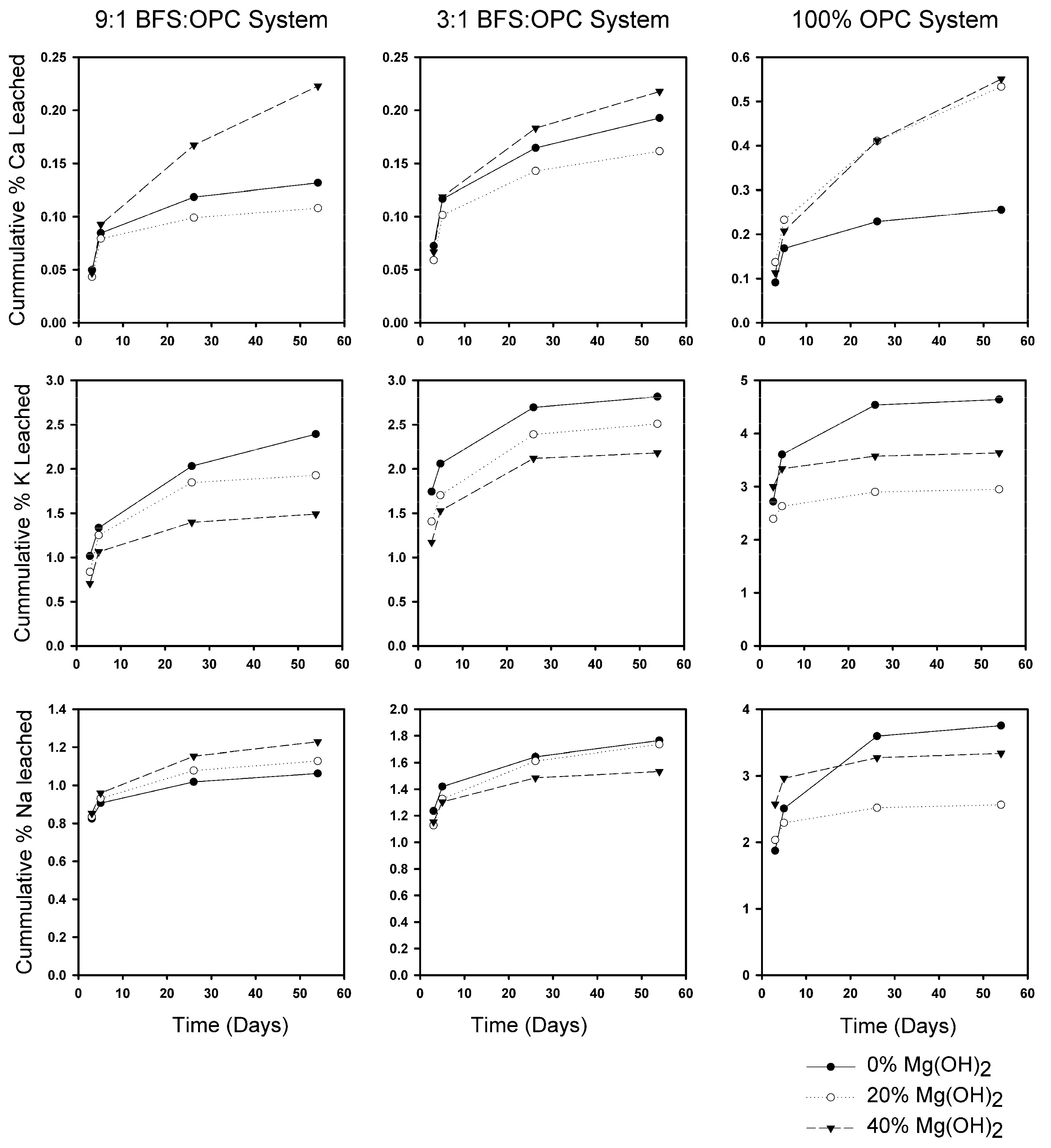
3.2. Leaching Behavior
3.2.1. OPC Versus BFS
3.2.2. Effect of Mg(OH)2 on Leaching
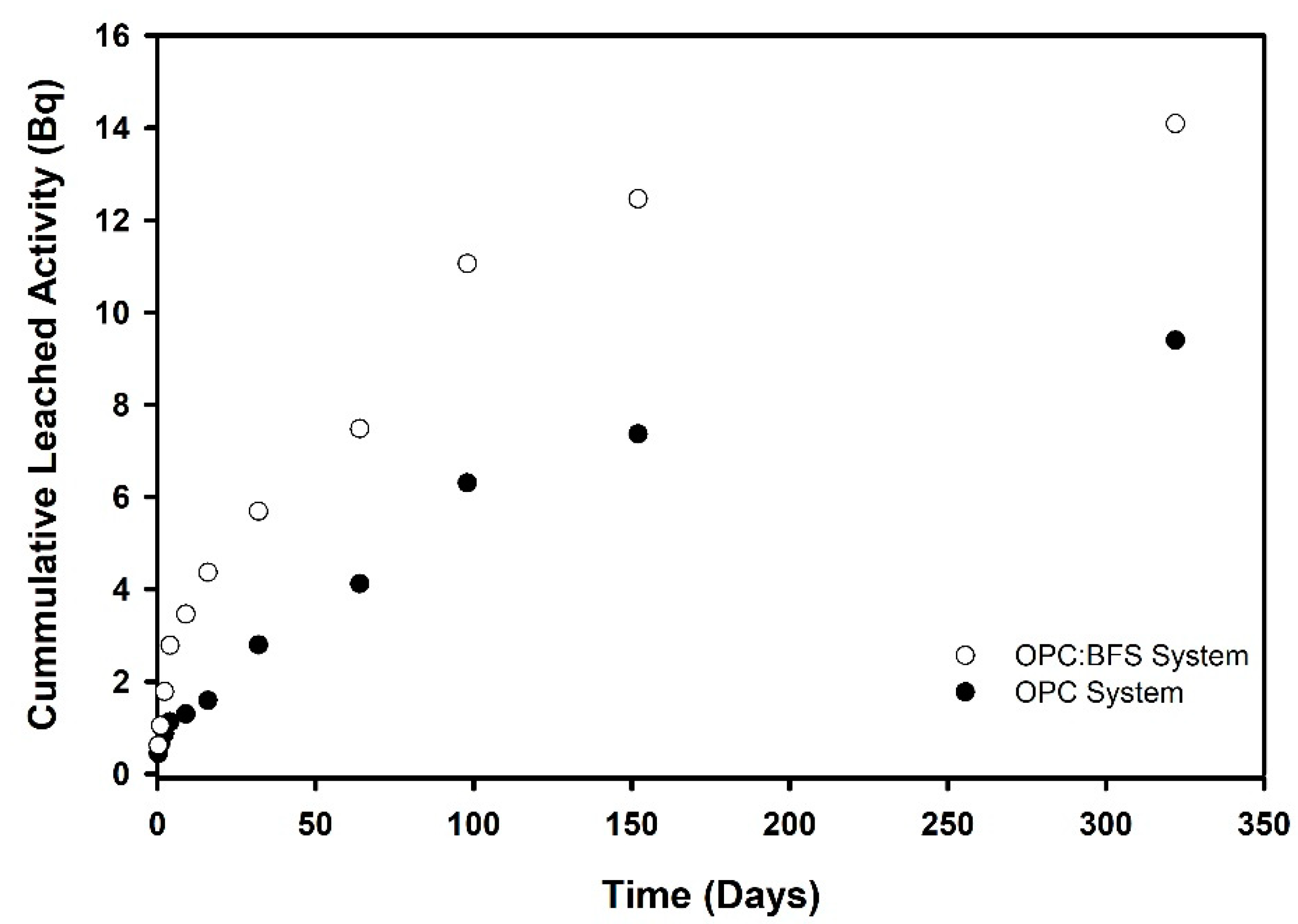
3.2.3. Leaching Rates
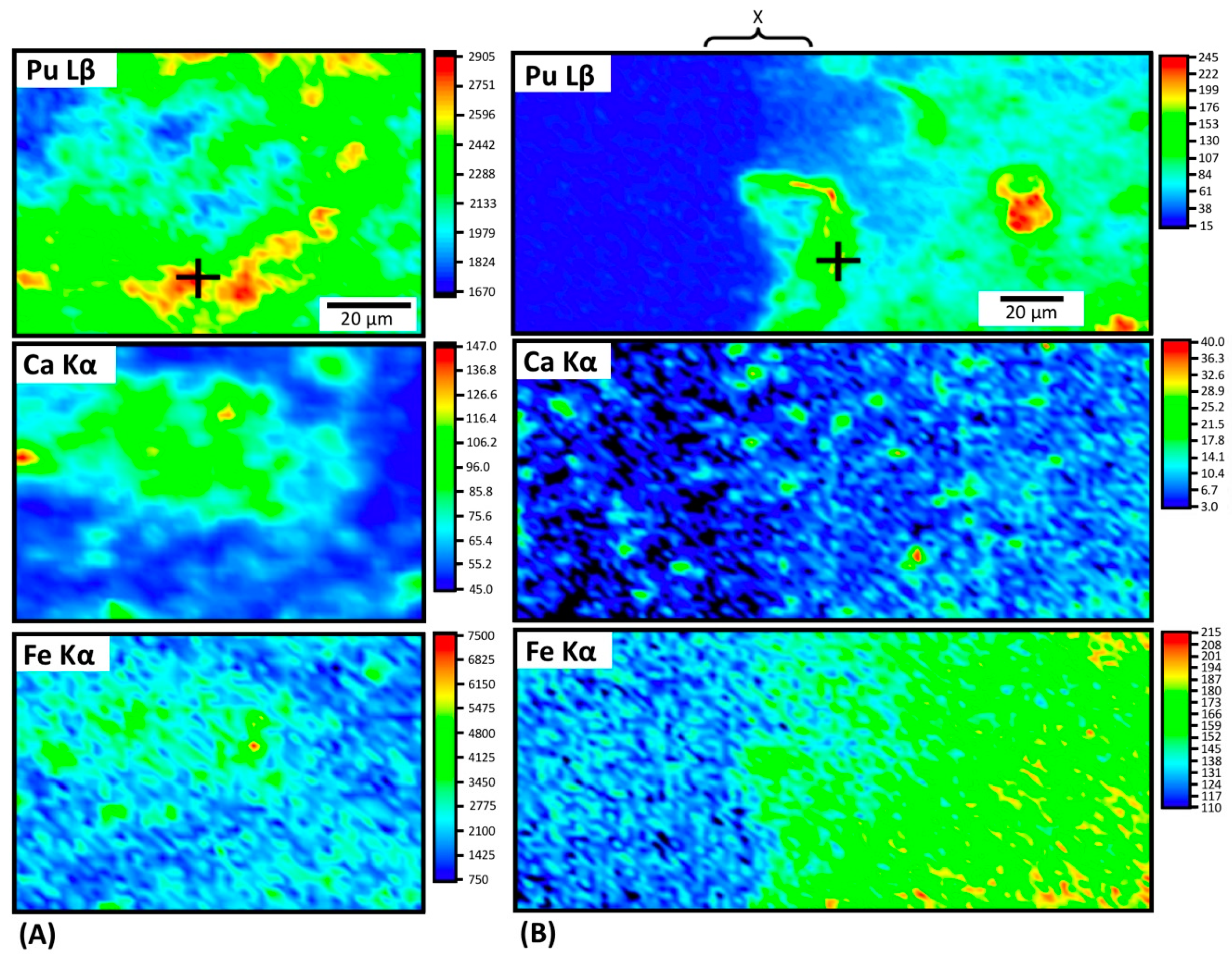
3.3. Characterization of Pu in Altered Cements—Plutonium Distribution and Speciation in Cements
X-Ray Fluorescence (XRF) Mapping
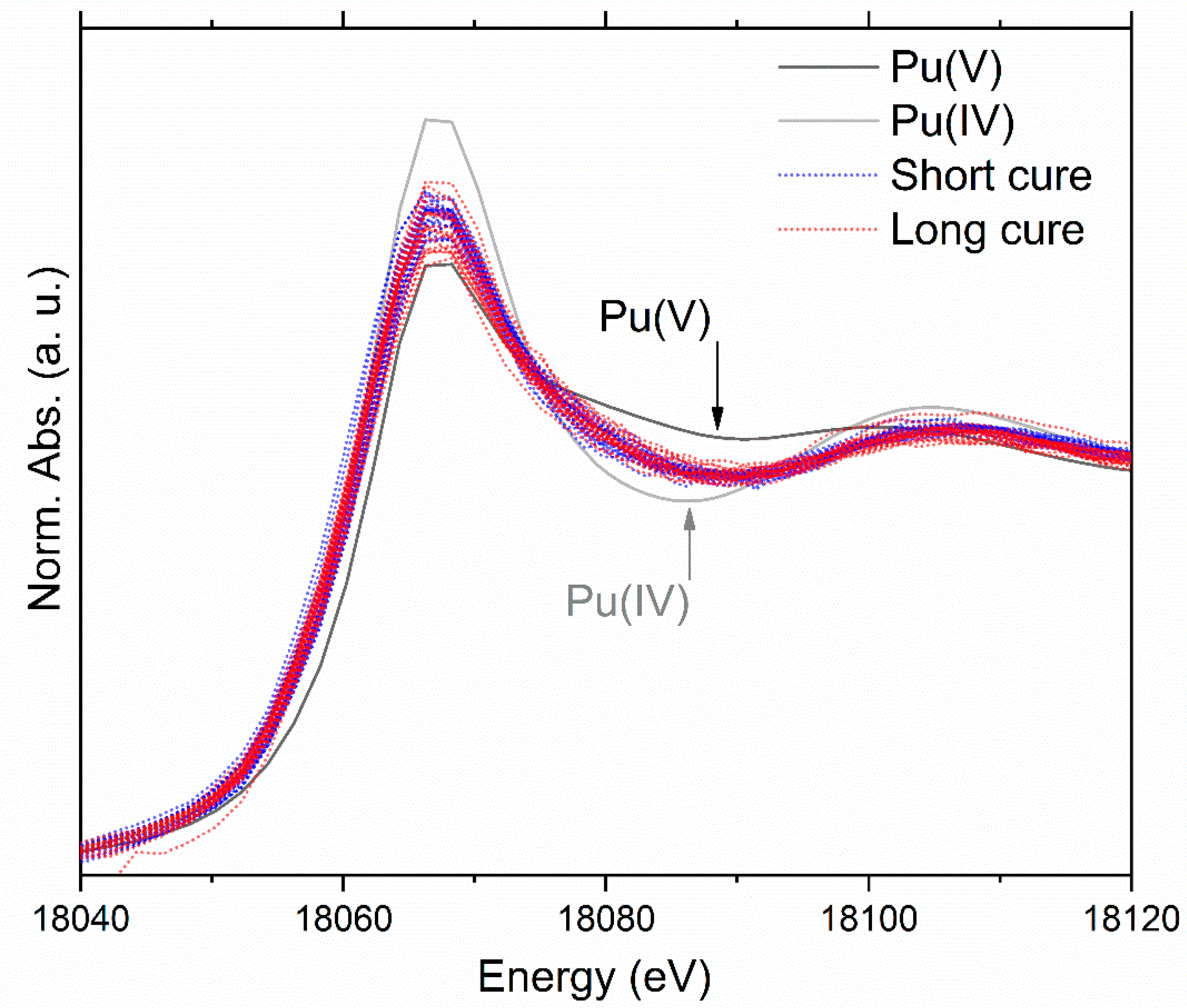
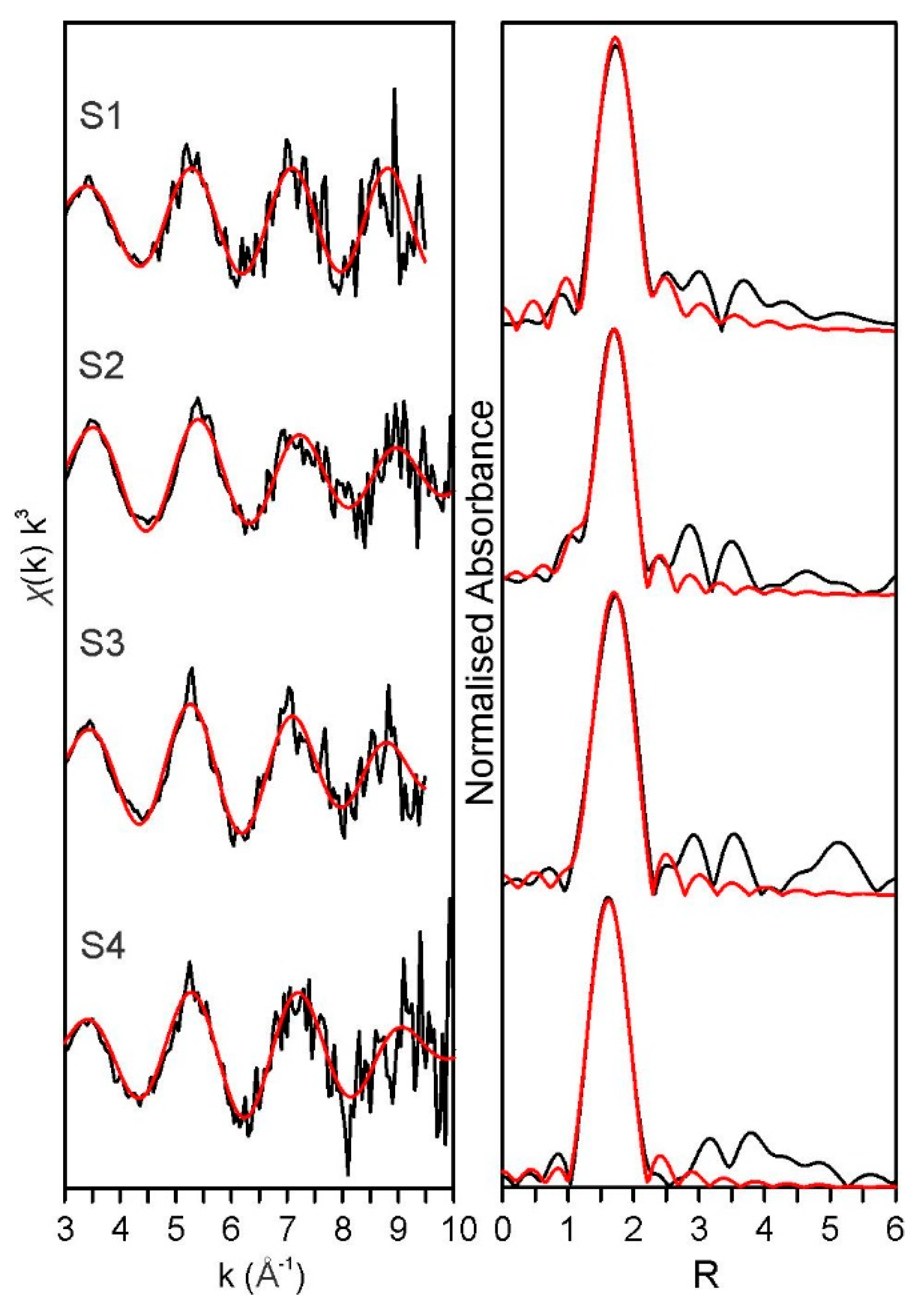
3.4. X-ray Absorption Spectroscopy Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, S.R.; Gray, J.; Smith, A.D. Discharges to the environment from the Sellafield site 1952–1992. J. Radiol. Prot. 1995, 15, 99–131. [Google Scholar]
- Gregson, C.R.; Goddard, D.T.; Sarsfield, M.J.; Taylor, R.J. Combined electron microscopy and vibrational spectroscopy study of corroded Magnox sludge from a legacy spent nuclear fuel storage pond. J. Nucl. Mat. 2011, 412, 145–156. [Google Scholar] [CrossRef]
- NDA; RWMD. The 2007 UK Radioactive Waste Inventory; NDA/RWMD/004; NDA: Moor Row, UK, March 2008.
- Gregson, C.R.; Hastings, J.J.; Sims, H.E.; Steele, H.M.; Taylor, R.J. Characterisation of plutonium species in alkaline liquors sampled from a UK legacy nuclear fuel storage pond. Anal. Methods 2011, 3, 1957–1968. [Google Scholar] [CrossRef]
- Nuclear Decommissioning Authority Strategy 2011; Nuclear Decommissioning Authority: Moor Row, UK, March 2011.
- Palmer, J.D.; Fairhall, G.A. Properties of cement systems containing intermediate level wastes. Cem. Concr. Res. 1992, 22, 325–330. [Google Scholar] [CrossRef]
- Managing Radioactive Waste Safely—A Framework for Implementing Geological Disposal; Department of Environment, Food and Rural Affairs: London, UK, 2008.
- Baldwin, T.; Chapman, N.; Neall, F. Geological Disposal Options for High-Level Waste and Spent Fuel; Nuclear Decommissioning Authority: Moor Row, UK, 2008.
- Chambers, A.; Gould, L.; Harris, A.; Pilkington, N.; Williams, S. Evolutions of the Near Field of NIREX Disposal Concept; Nirex: Harwell, Oxfordshire, UK, 2003. [Google Scholar]
- Generic Repository Studies—Generic Post Closure Performance Assessment; Report N/080; Nirex: Harwell, Oxfordshire, UK, 2003.
- Generic Repository Studies—Generic Waste Package Specification; Volume 1—Specification; Report N/104; Nirex: Harwell, Oxfordshire, UK, 2007.
- NDA. Waste Package Specification and Guidance Documentation. WPS/906: Guidance on the Packaging of Closed Sources; Number 10157950; NDA: Moor Row, UK, February 2009.
- Clark, D.L.; Hecker, S.S.; Jarvinen, G.D.; Neu, M.P. Plutonium. In The Chemistry of the Actinide and Transactinide Elements, 4th ed.; Morss, L.R., Edlestien, N.M., Fruger, J., Eds.; Springer: Dordrecht, The Netherlands, 2010; Volume 2, pp. 813–1264. [Google Scholar]
- Leaching Characteristics of Moulded or Monolithic Building and Waste Materials: Determination of Leaching of Inorganic Components with the Diffusion Test; EA NEN 7375; UK Environment Agency: Bristol, UK, 2005.
- Heald, S.M.; Cross, J.O.; Brewe, D.J.; Gordon, R.A. The PNC/XOR X-ray microprobe station at APS sector 20. Nucl. Instrum. Methods Phys. Res. A 2007, 582, 215–217. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Syncotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Di Giandomenico, M.V.; Le Naour, C.; Simoni, E.; Guillaumont, D.; Moisy, P.; Hennig, C.; Conradson, S.D.; Den Auwer, C. Structure of early actinides (V) in acidic solutions. Radiochim. Acta 2009, 97, 347–353. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of x-ray absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Caldwell, R.J.; Rawlinson, S.; Butcher, E.J.; Godfrey, L.H. Characterisation of full-scale historic inactive cement-based intermediate level nuclear wasteforms. In Stabilisation/Solidification Treatment and Remediation, Advances for S/S in Waste Contaminated Land; Al-Tabbaa, A., Stegemann, J.A., Eds.; Taylor and Francis: London, UK, 2005; pp. 139–148. [Google Scholar]
- Collier, N.C.; Milestone, N.B. The encapsulation of Mg(OH)2 sludge in composite cement. Cem. Concr. Res. 2010, 40, 452–459. [Google Scholar] [CrossRef]
- Jain, J.; Neithalath, N. Analysis of calcium leaching behavior of plain and modified cement pastes in pure water. Cem. Concr. Comp. 2009, 31, 176–185. [Google Scholar] [CrossRef]
- Gorce, J.-P.; Milestone, N.B. Probing the microstructure and water phases in composite cement blendes. Cem. Concr. Res. 2007, 37, 310–318. [Google Scholar] [CrossRef]
- Neck, V.; Kim, J.I. Solubility and hydrolysis of tetravalent actinides. Radiochim. Acta 2001, 89, 1–16. [Google Scholar] [CrossRef]
- Schwotzer, M.; Scherer, T.; Gerdes, A. Protective or damage promoting effect of calcium carbonate layers on the surface of cement based materials in aqueous environments. Cem. Conc. Res. 2010, 40, 1410–1418. [Google Scholar] [CrossRef]
- Dow, C.; Glasser, F.P. Calcium carbonate efflorescence on Portland cement and building materials. Cem. Conc. Res. 2003, 33, 147–154. [Google Scholar] [CrossRef]
- Dardenne, K.; Seibert, A.; Denecke, M.A.; Marcquardt, C.M. Plutonium (III, IV, VI) speciation in Gorleben groundwater using XAFS. Radiochim. Acta 2009, 97, 91–97. [Google Scholar] [CrossRef]
- LoPresti, V.; Conradson, S.D.; Clark, D.L. XANES identification of plutonium speciation in RFETS samples. J. Alloy. Compd. 2007, 444–445, 540–543. [Google Scholar] [CrossRef]
- Rossberg, A.; Scheinost, A.C.; Schmeisser, N.; Rothe, J.; Kaden, P.; Schild, D.; Wiss, T.; Daehn, R. AcReDaS, an Actinide Reference Database for XAS, EELS, IR, Raman and NMR Spectroscopy. Available online: https://www.hzdr.de/acredas (accessed on 30 November 2018).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sample ID | Pu Spike | Cure Time (Days) | Leached | Spectroscopy |
|---|---|---|---|---|---|
| BFS/OPC Short cure | A | 100 kBq 239Pu | 13 | Yes—322 days | No |
| BFS/OPC Long cure | B | 100 kBq 239Pu | 130 | Yes—322 days | No |
| OPC Short cure | C | 100 kBq 239Pu | 13 | Yes—322 days | No |
| OPC Long cure | D | 100 kBq 239Pu | 130 | Yes—322 days | No |
| OPC Short cure | 1 | 460 kBq 242Pu | 13 | No | Yes |
| OPC Short cure | 2 | 460 kBq 242Pu | 13 | Yes—98 days | Yes |
| OPC Long cure | 3 | 460 kBq 242Pu | 130 | Yes—64 days | Yes |
| OPC Long cure | 4 | 460 kBq 242Pu | 130 | No | Yes |
| Sample | Sample ID | Initial Leach Rate % Pu per Day | Long Term Leach Rate % Pu per Day |
|---|---|---|---|
| BFS/OPC Short Cure | A | 6.0 × 10−4 | 1.8 × 10−5 |
| BFS/OPC Long Cure | B | 1.0 × 10−3 | 2.9 × 10−5 |
| OPC Short Cure | C | 6.5 × 10−4 | 2.9 × 10−5 |
| OPC Long Cure | D | 1.0 × 10−3 | 4.4 × 10−5 |
| Sample | Path | S02 | Error ± | N | R | Error ± | σ2 | Error ± | r-Factor |
|---|---|---|---|---|---|---|---|---|---|
| S1 (4) | Pu—Oeq | 0.80 | 0.10 | 5.5 * | 2.27 | 0.01 | 0.002 | 0.002 | 0.009 |
| S2 (3) | Pu—Oeq | 0.93 | 0.11 | 6.0 * | 2.25 | 0.02 | 0.009 | 0.003 | 0.013 |
| S3 (4) | Pu—Oax | 0.90 * | 0.6 * | 1.81 | 0.04 | 0.002 | 0.007 | 0.017 | |
| Pu—Oeq | 6.0 * | 2.29 | 0.02 | 0.006 | 0.001 | ||||
| S4 (3) | Pu—Oax | 0.90 * | 0.7 * | 1.8 | 0.01 | 0.002 | 0.003 | 0.008 | |
| Pu—Oeq | 6.0 * | 2.25 | 0.02 | 0.009 | 0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Law, K.A.; Parry, S.; Bryan, N.D.; Heath, S.L.; Heald, S.M.; Knight, D.; O’Brien, L.; Fuller, A.J.; Bower, W.R.; Law, G.T.W.; et al. Plutonium Migration during the Leaching of Cemented Radioactive Waste Sludges. Geosciences 2019, 9, 31. https://doi.org/10.3390/geosciences9010031
Law KA, Parry S, Bryan ND, Heath SL, Heald SM, Knight D, O’Brien L, Fuller AJ, Bower WR, Law GTW, et al. Plutonium Migration during the Leaching of Cemented Radioactive Waste Sludges. Geosciences. 2019; 9(1):31. https://doi.org/10.3390/geosciences9010031
Chicago/Turabian StyleLaw, Kathleen A., Stephen Parry, Nicholas D. Bryan, Sarah L. Heath, Steven M. Heald, Darrell Knight, Luke O’Brien, Adam J. Fuller, William R. Bower, Gareth T. W. Law, and et al. 2019. "Plutonium Migration during the Leaching of Cemented Radioactive Waste Sludges" Geosciences 9, no. 1: 31. https://doi.org/10.3390/geosciences9010031
APA StyleLaw, K. A., Parry, S., Bryan, N. D., Heath, S. L., Heald, S. M., Knight, D., O’Brien, L., Fuller, A. J., Bower, W. R., Law, G. T. W., & Livens, F. R. (2019). Plutonium Migration during the Leaching of Cemented Radioactive Waste Sludges. Geosciences, 9(1), 31. https://doi.org/10.3390/geosciences9010031