Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation



Abstract
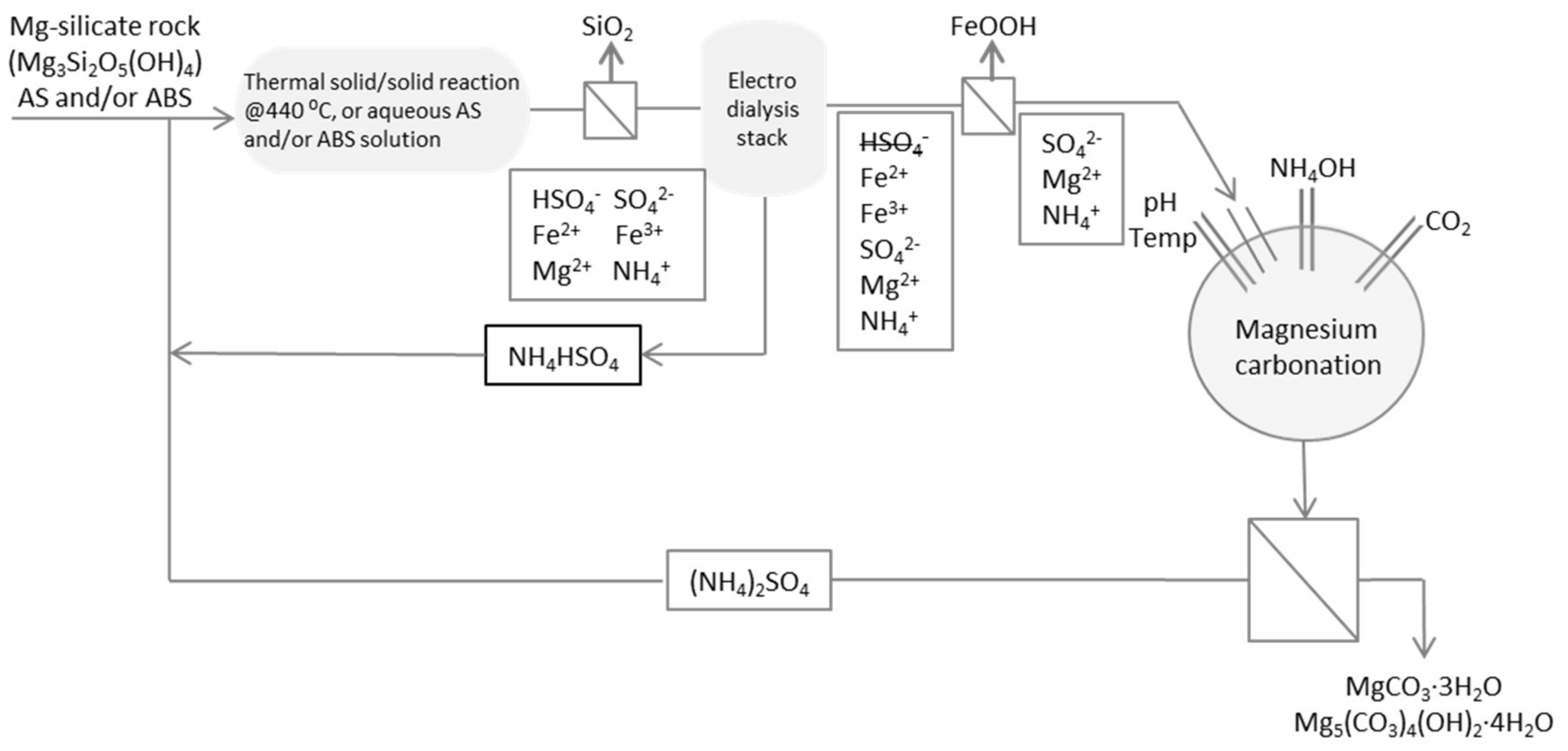
:1. Introduction
1.1. Electrodialysis and Sulfate Separation
1.2. Exergy Calculations
2. Materials and Methods
2.1. Material
2.2. Methods
2.2.1. Experimental Procedures
2.2.2. Analysis Methods and Devices, Calibration Curves
3. Results and Discussion
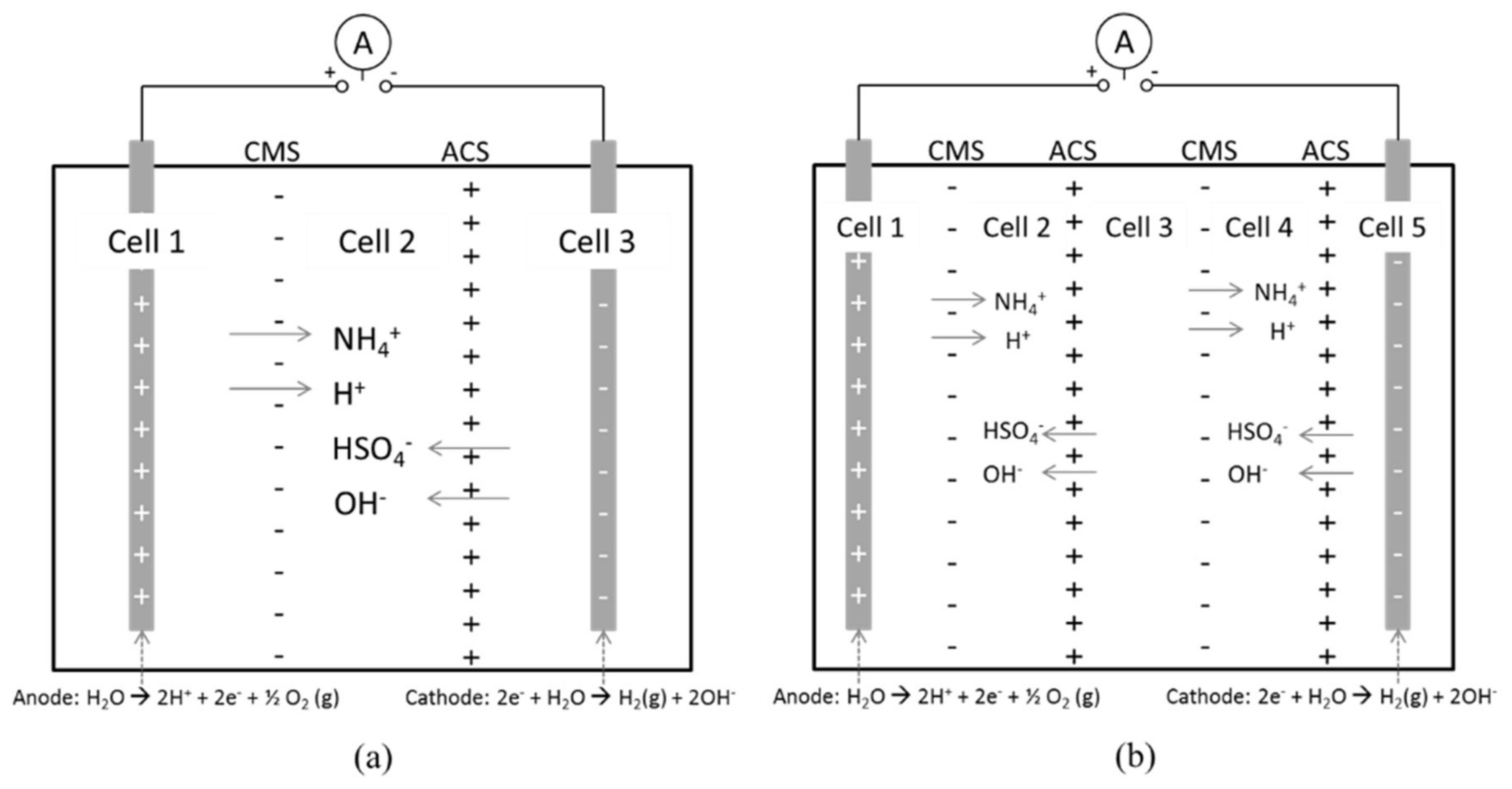
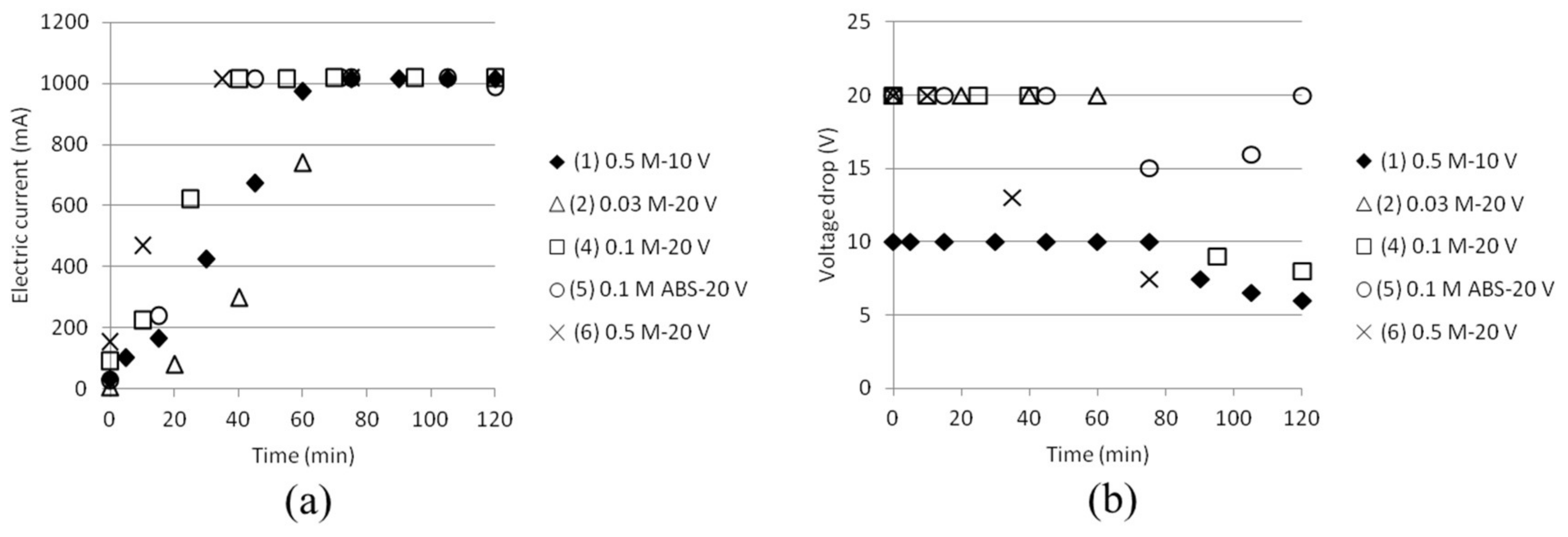
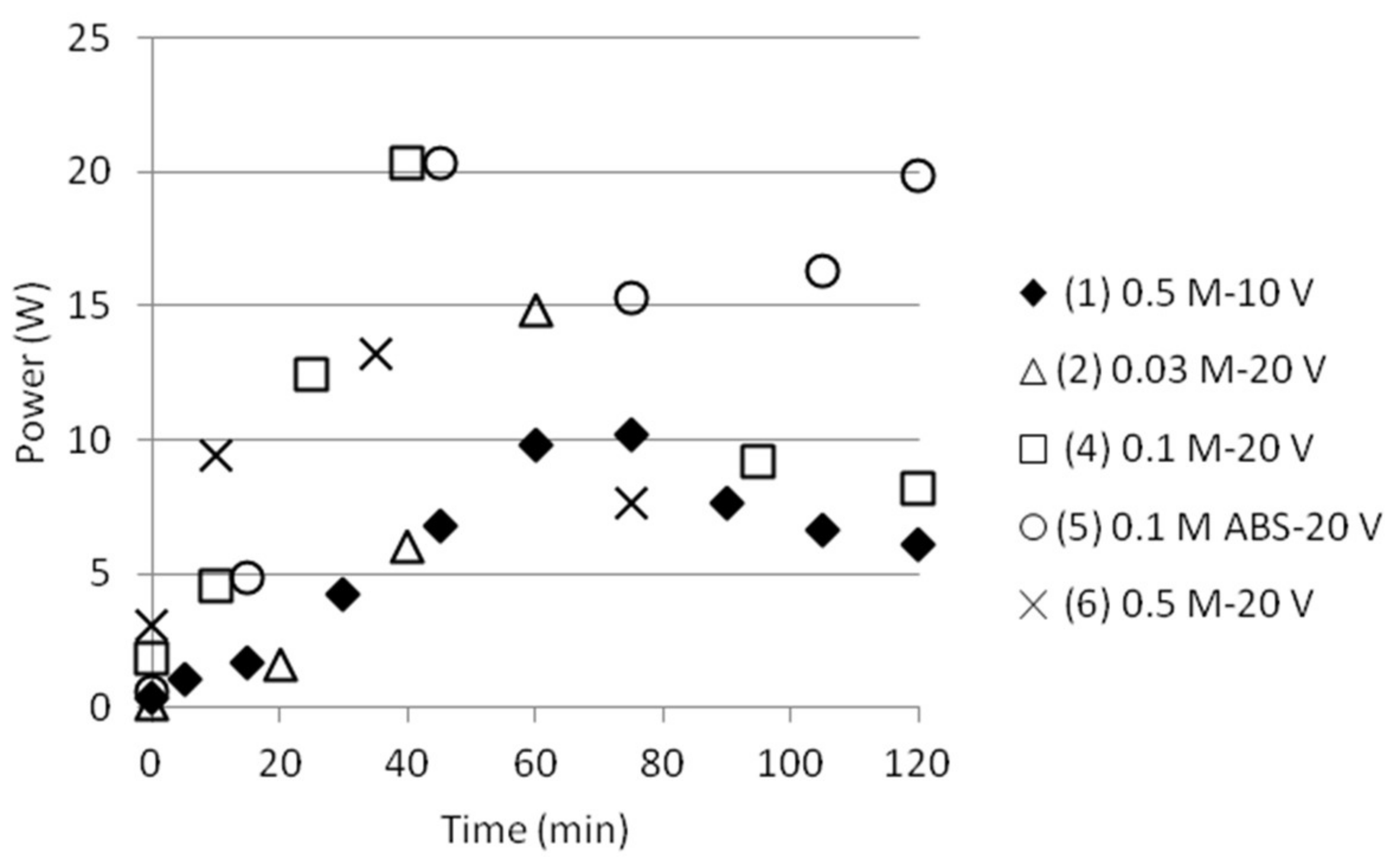
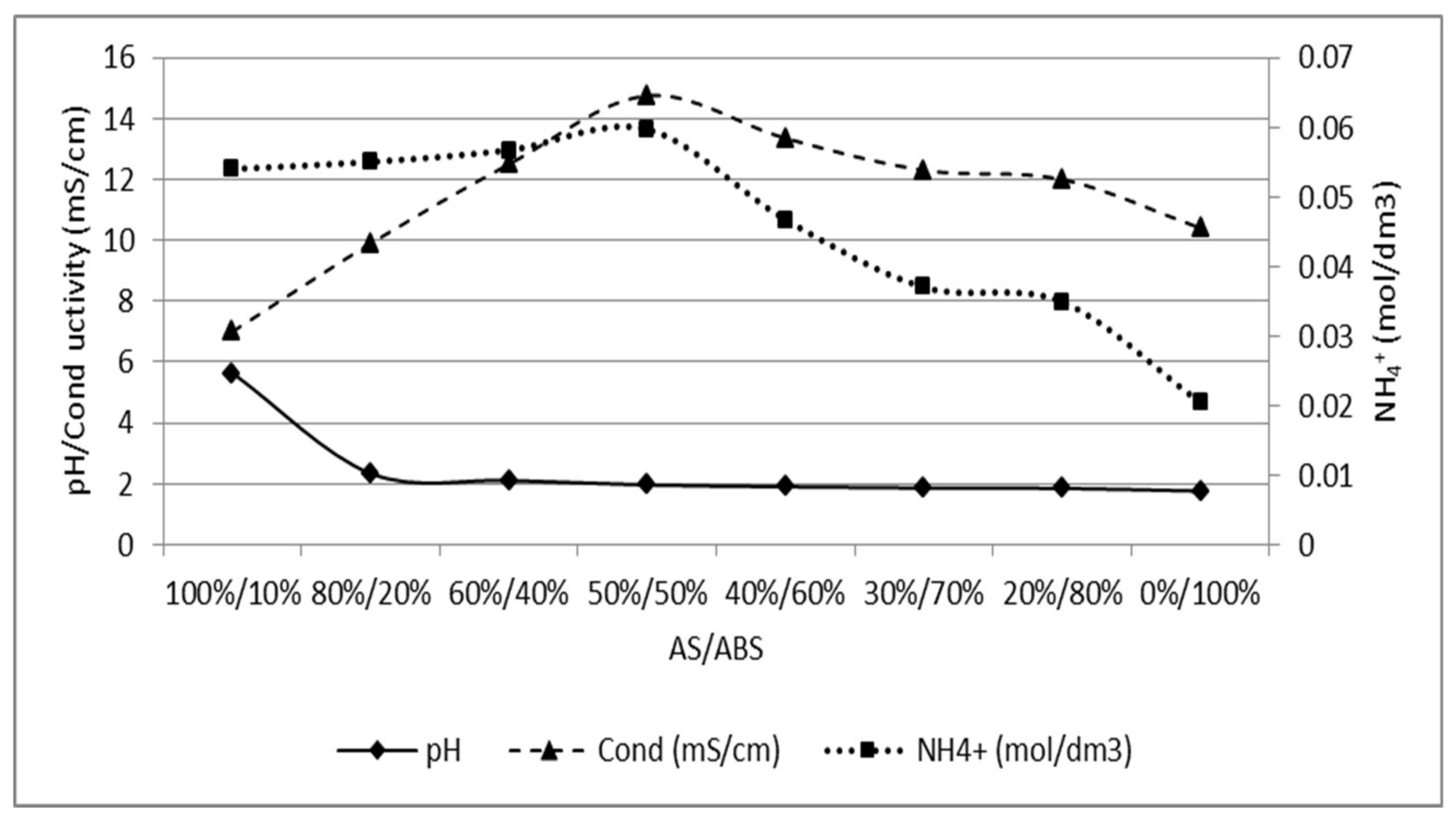
3.1. Reference State of the Electrodialysis Stack
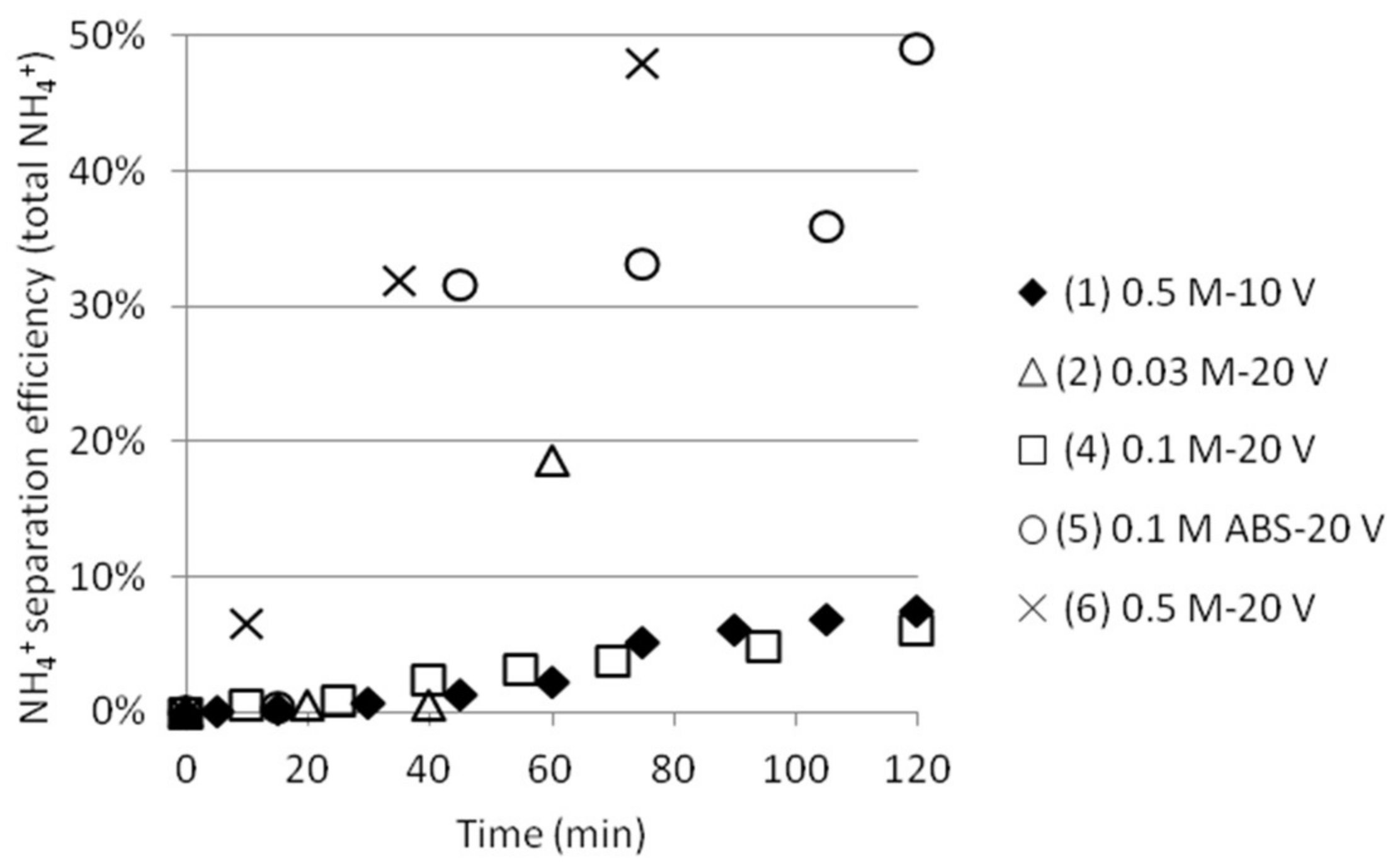
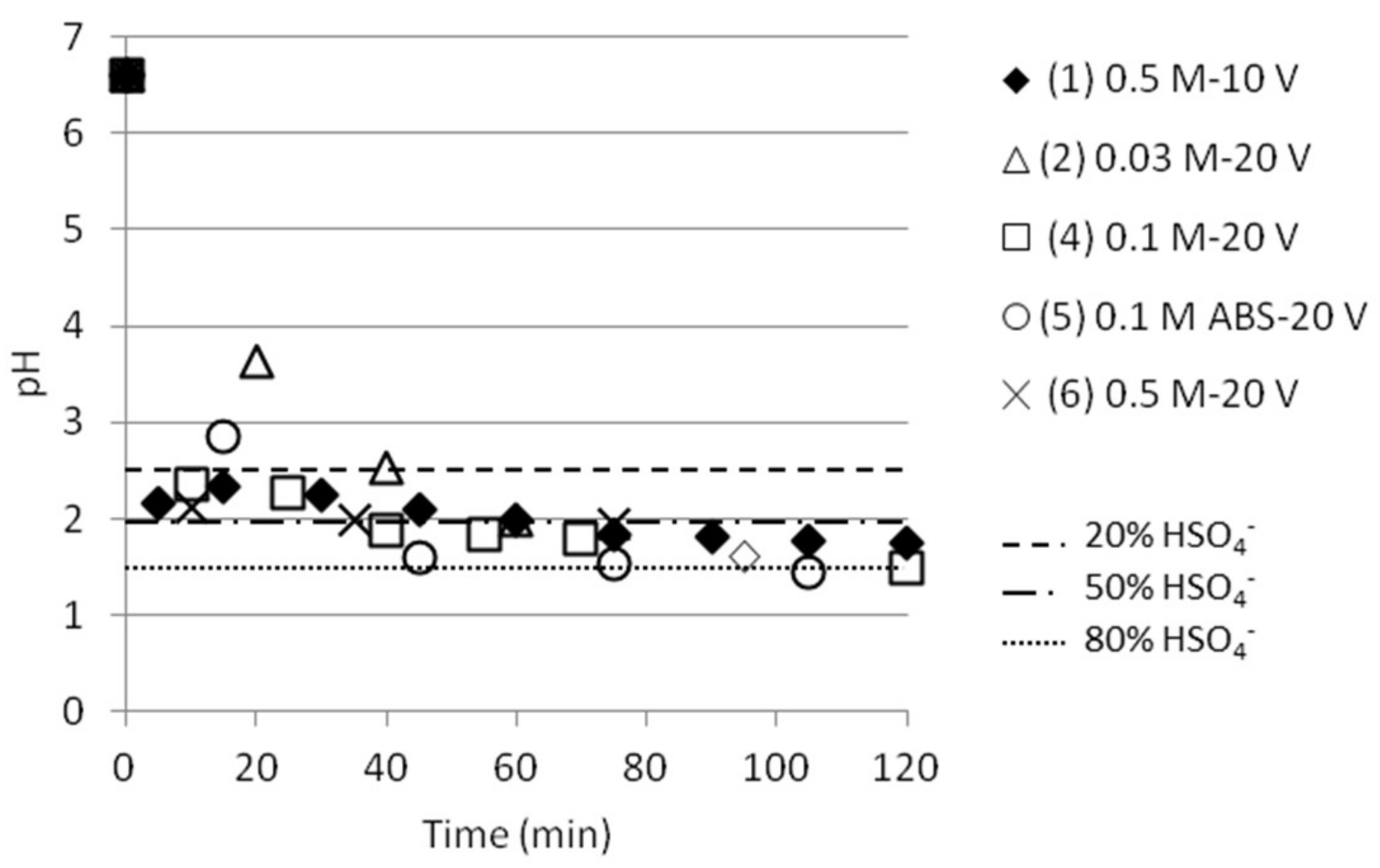
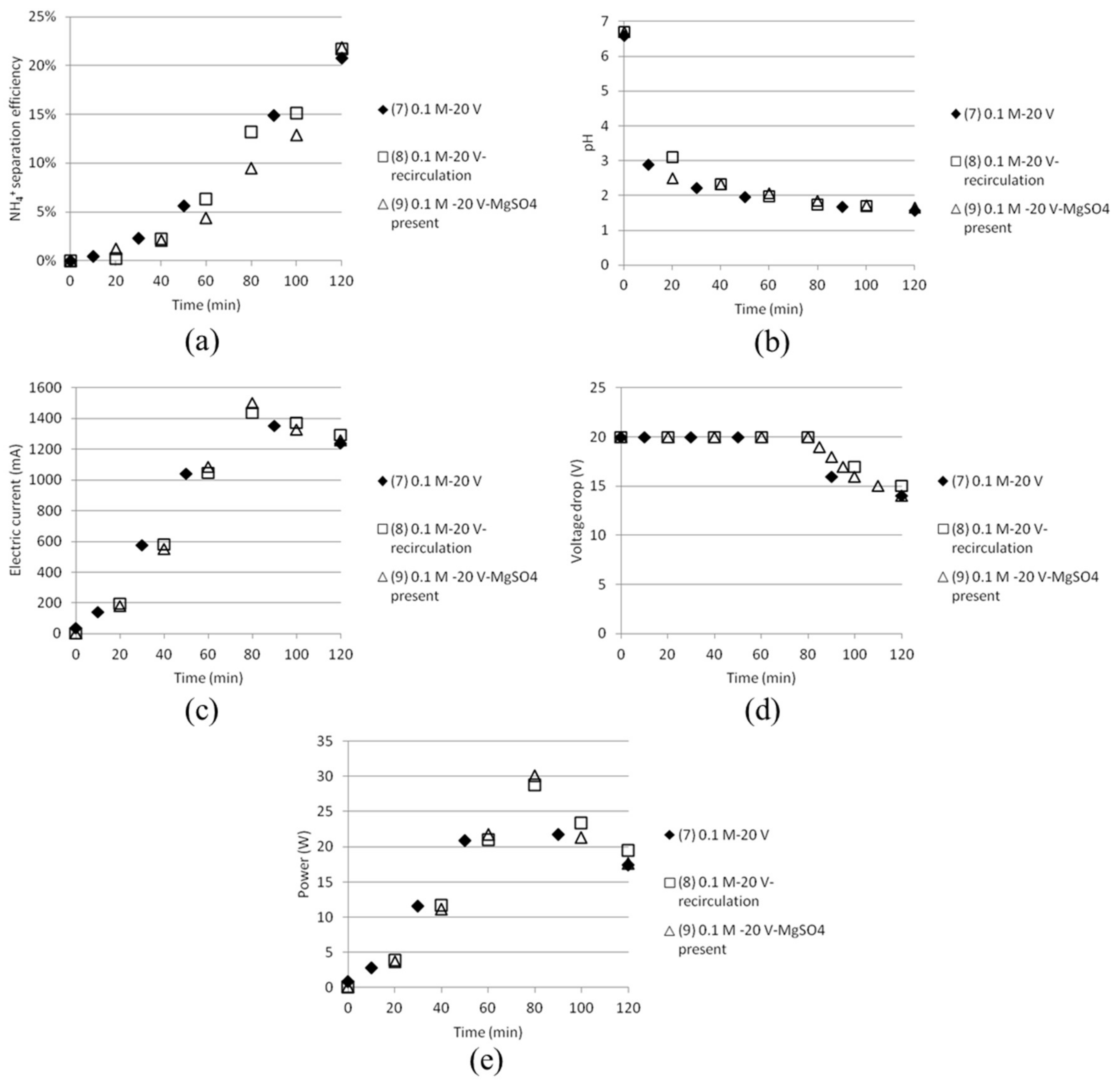
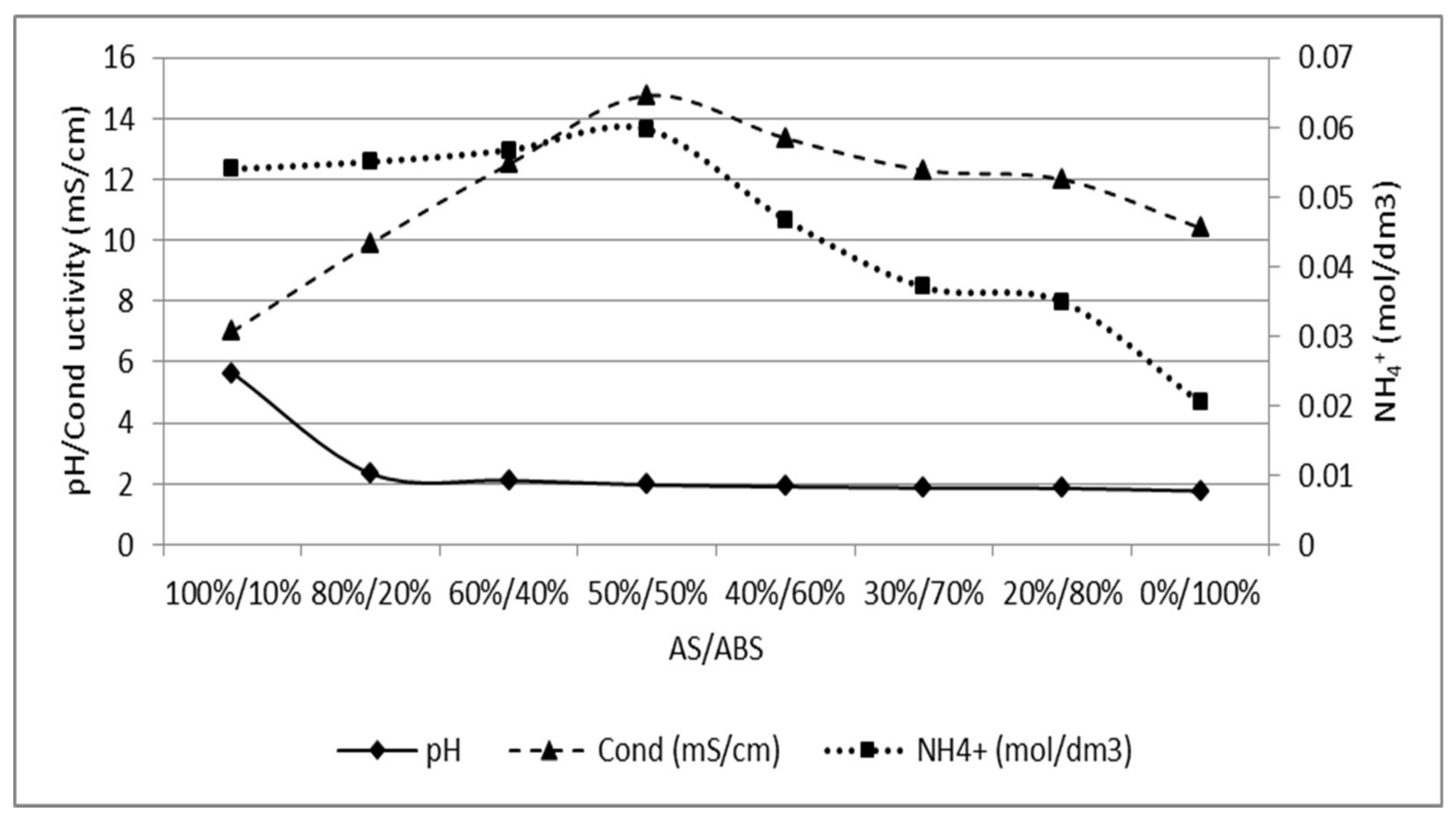
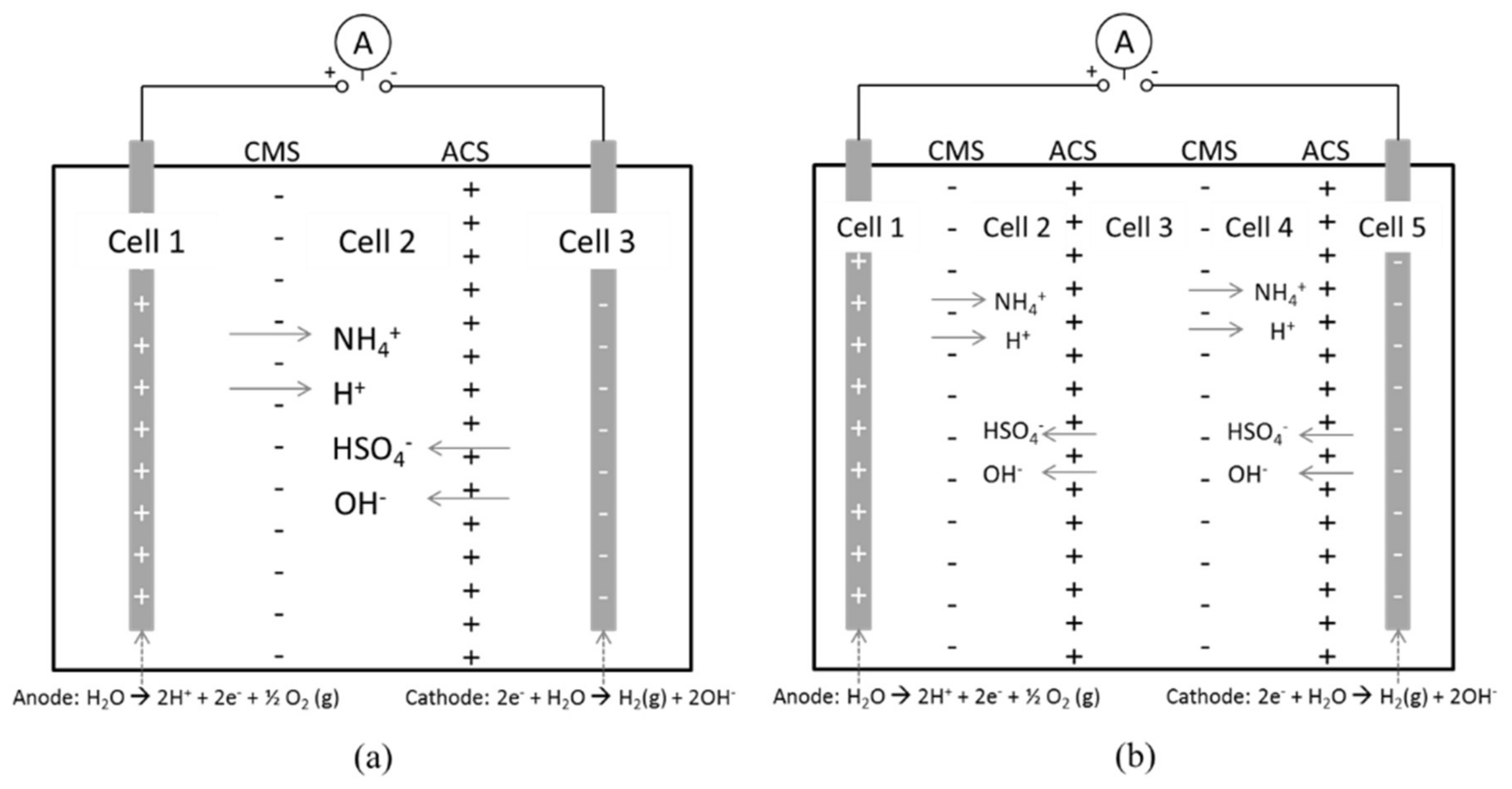
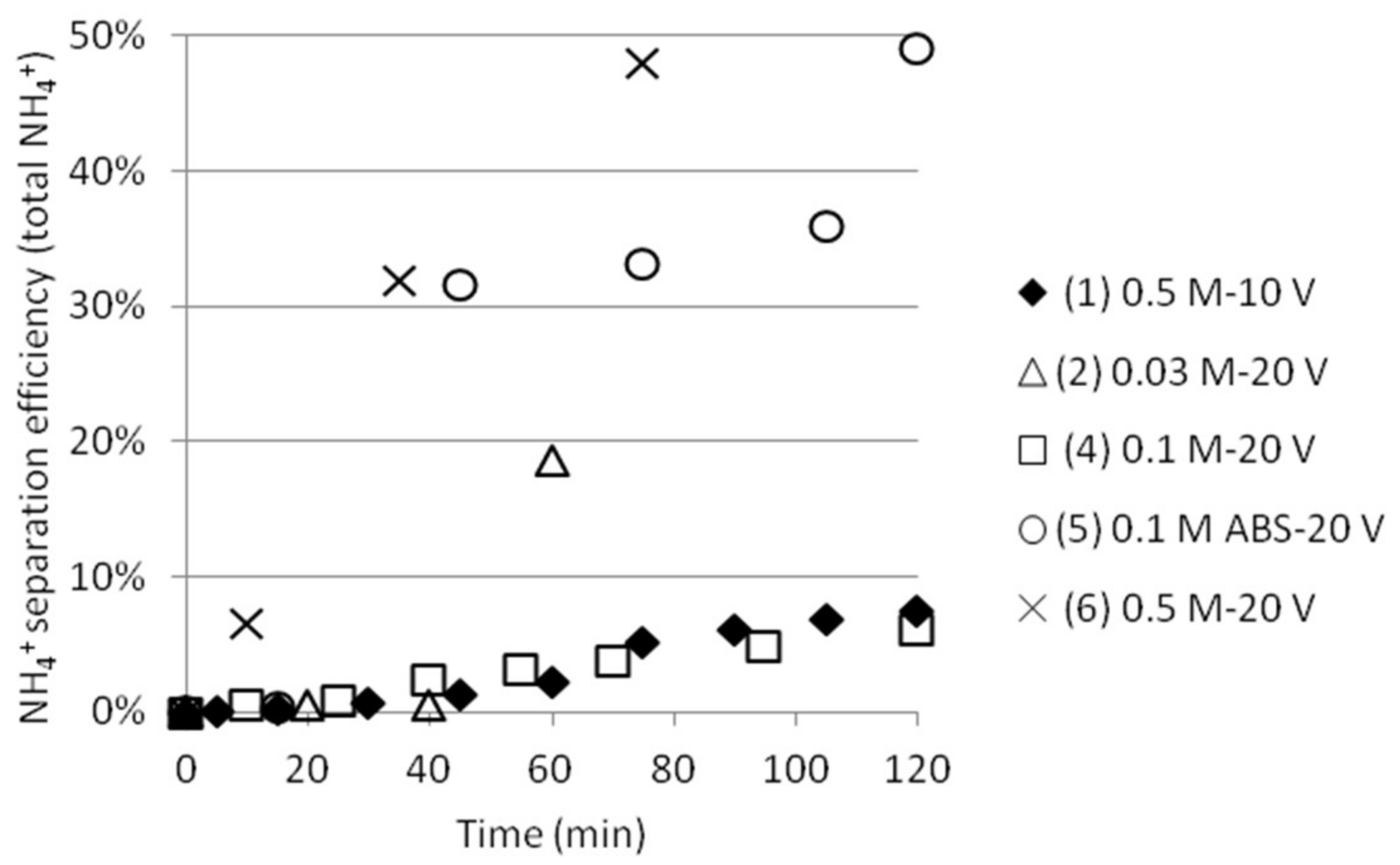
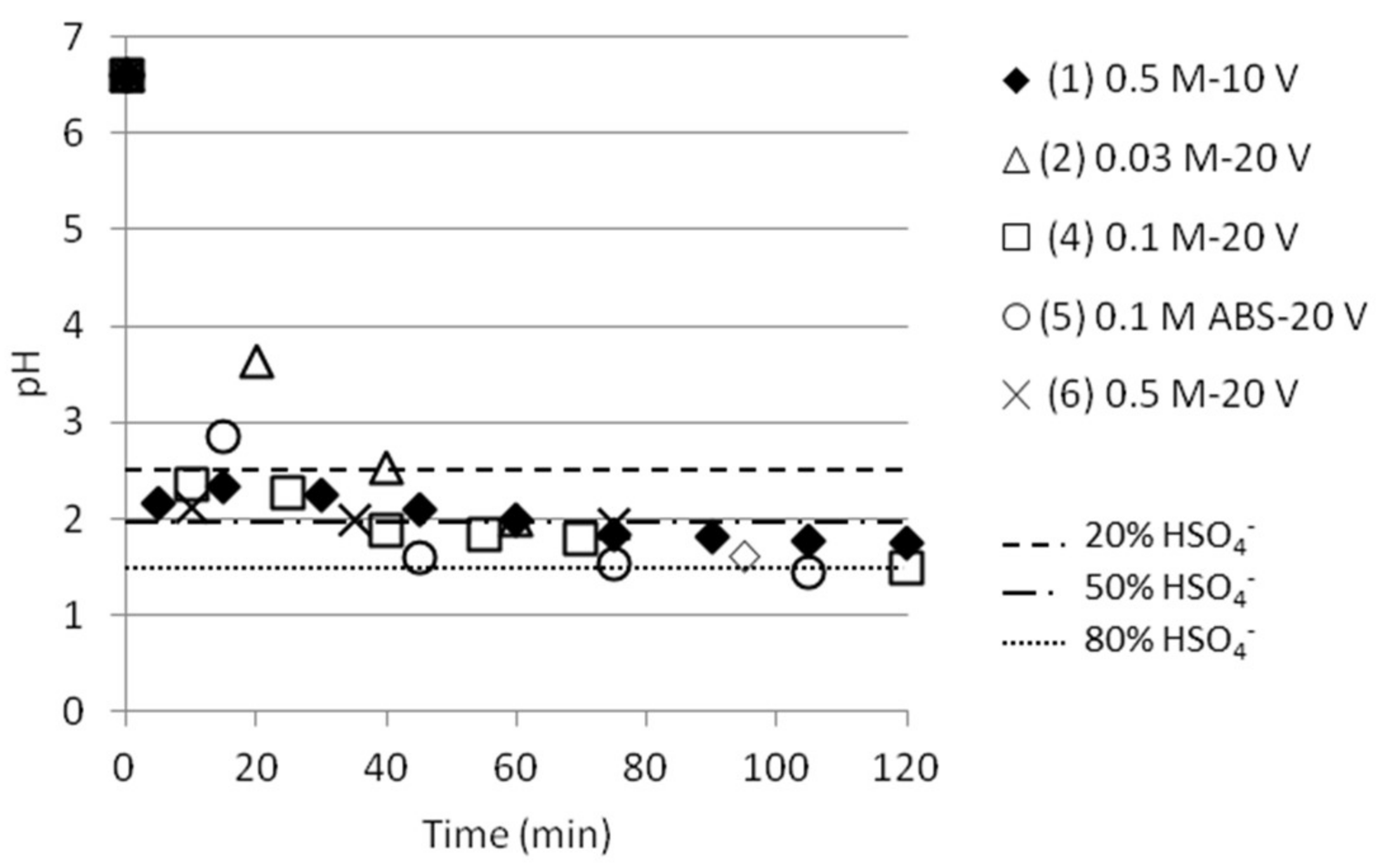
3.2. Separation of ABS with a Three Compartment Setup
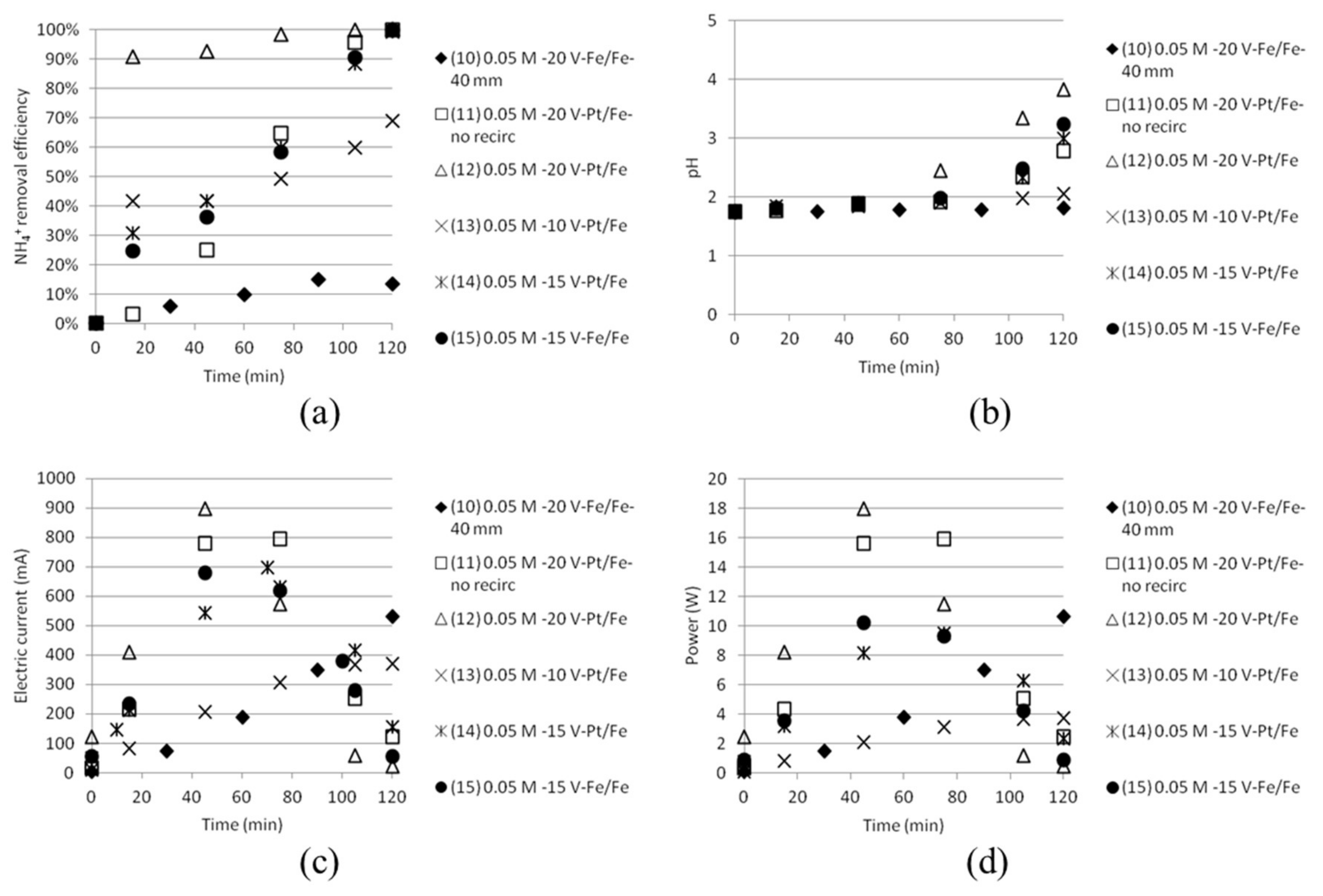
3.3. Separation of ABS in Double Distance Anode and Cathode Cells
3.4. Separation of ABS with a Five Compartment Setup
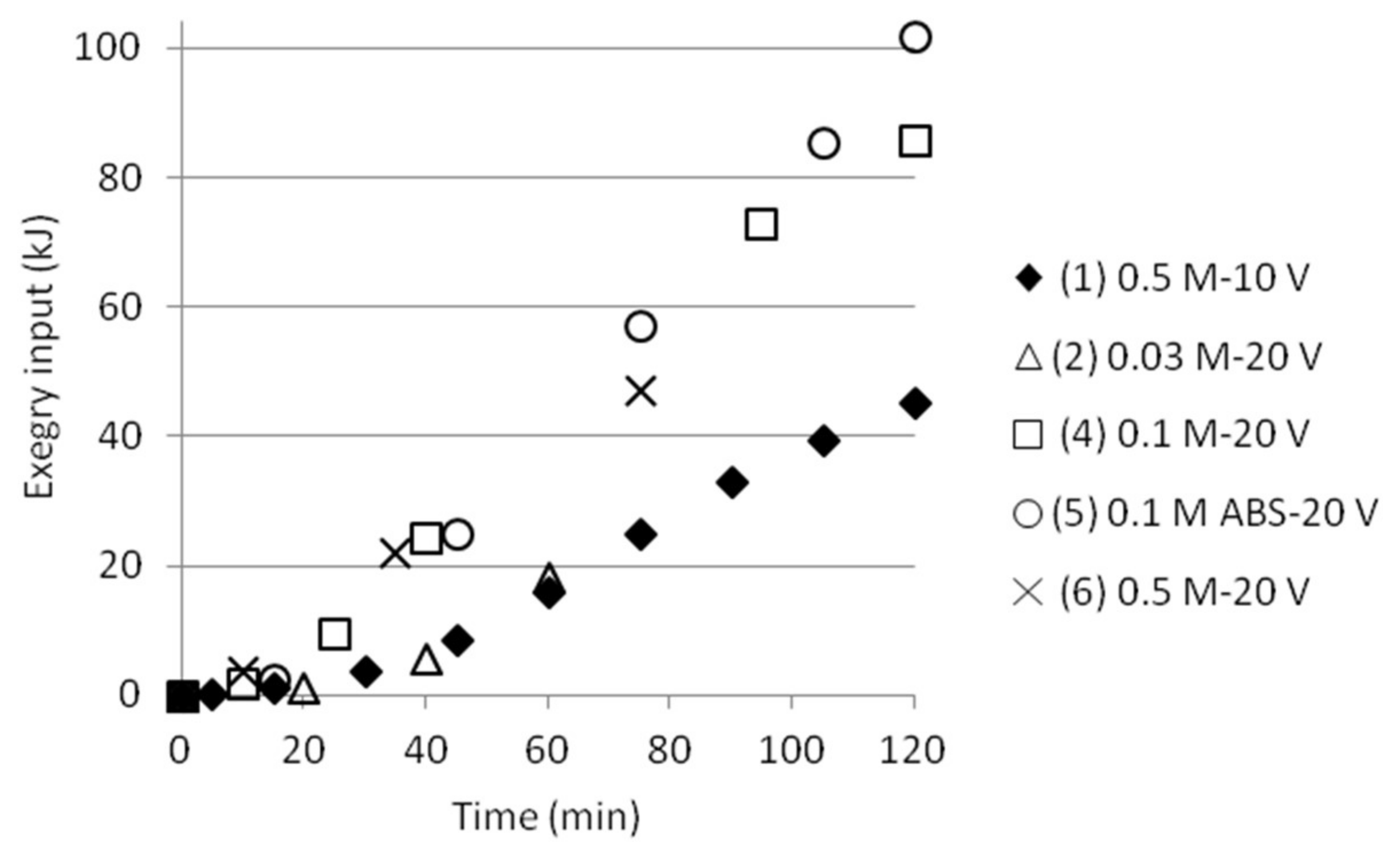
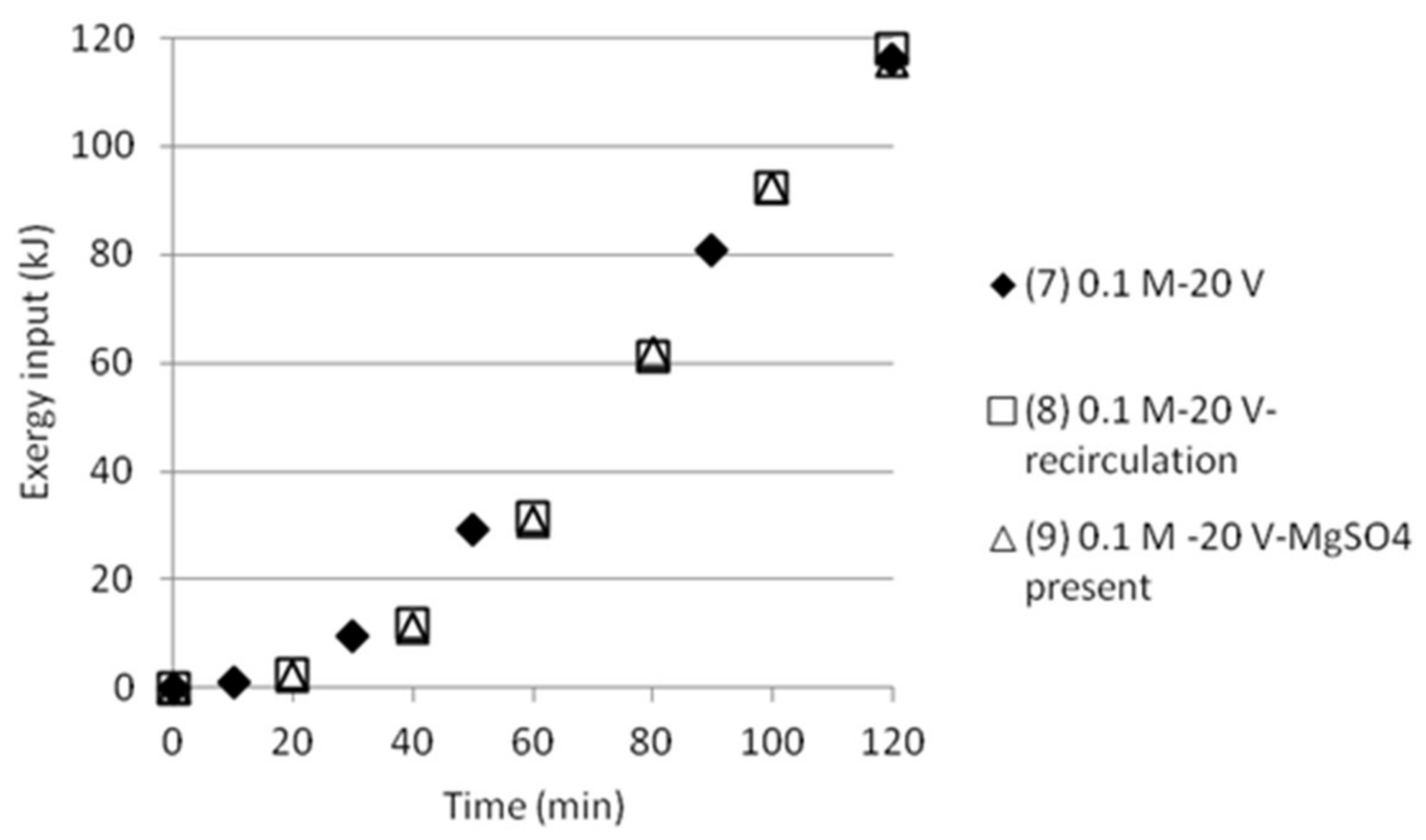
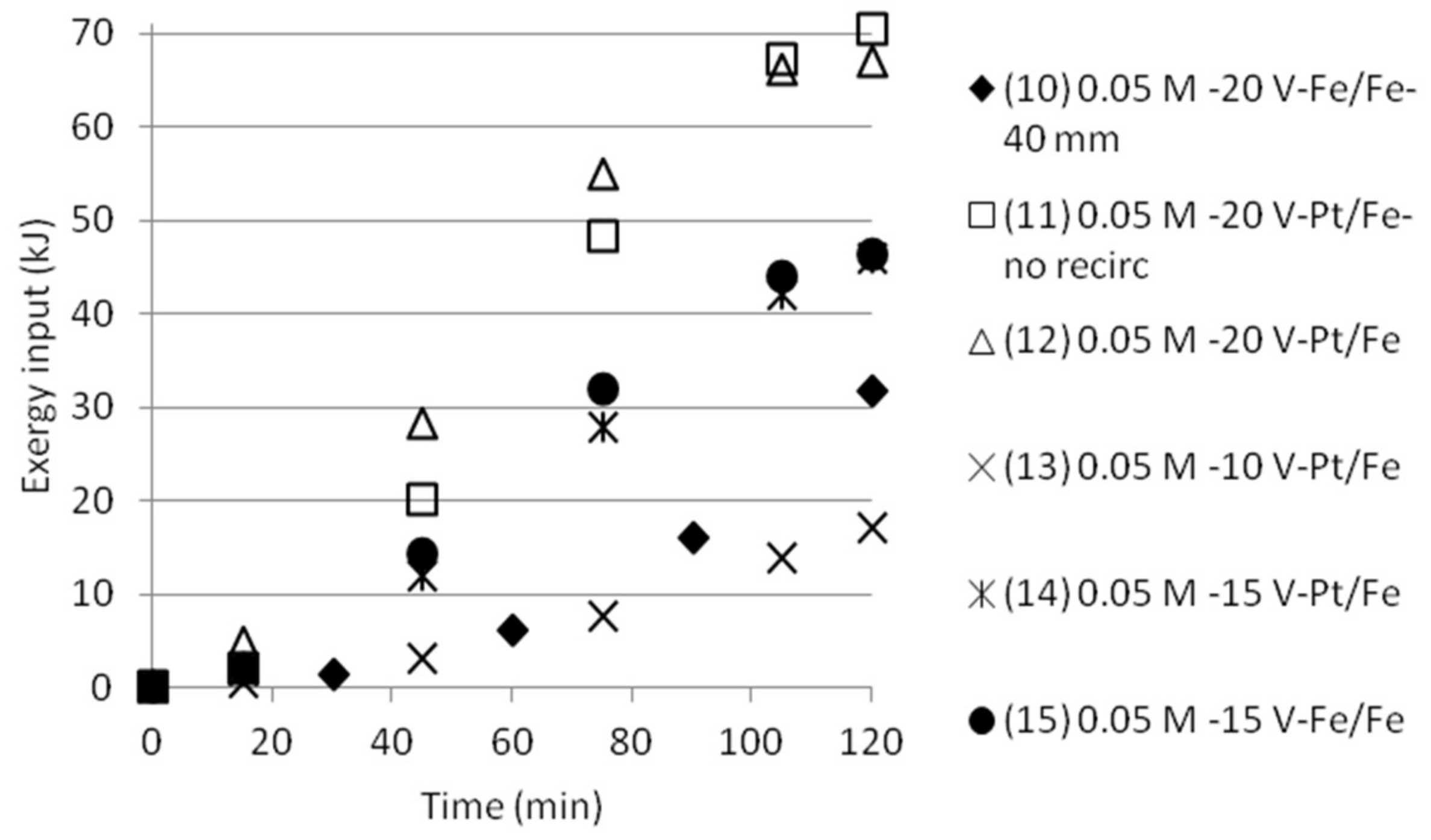
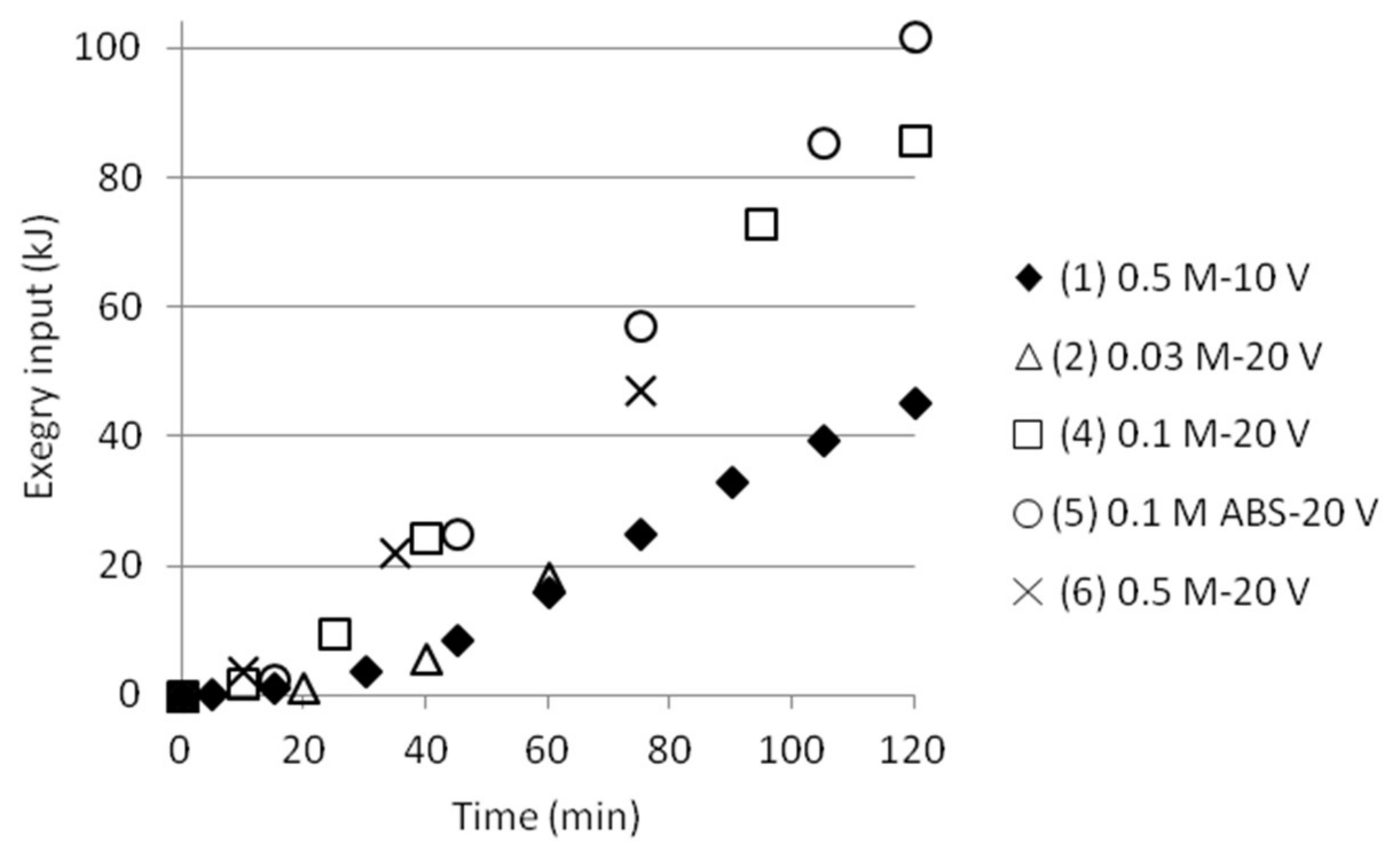
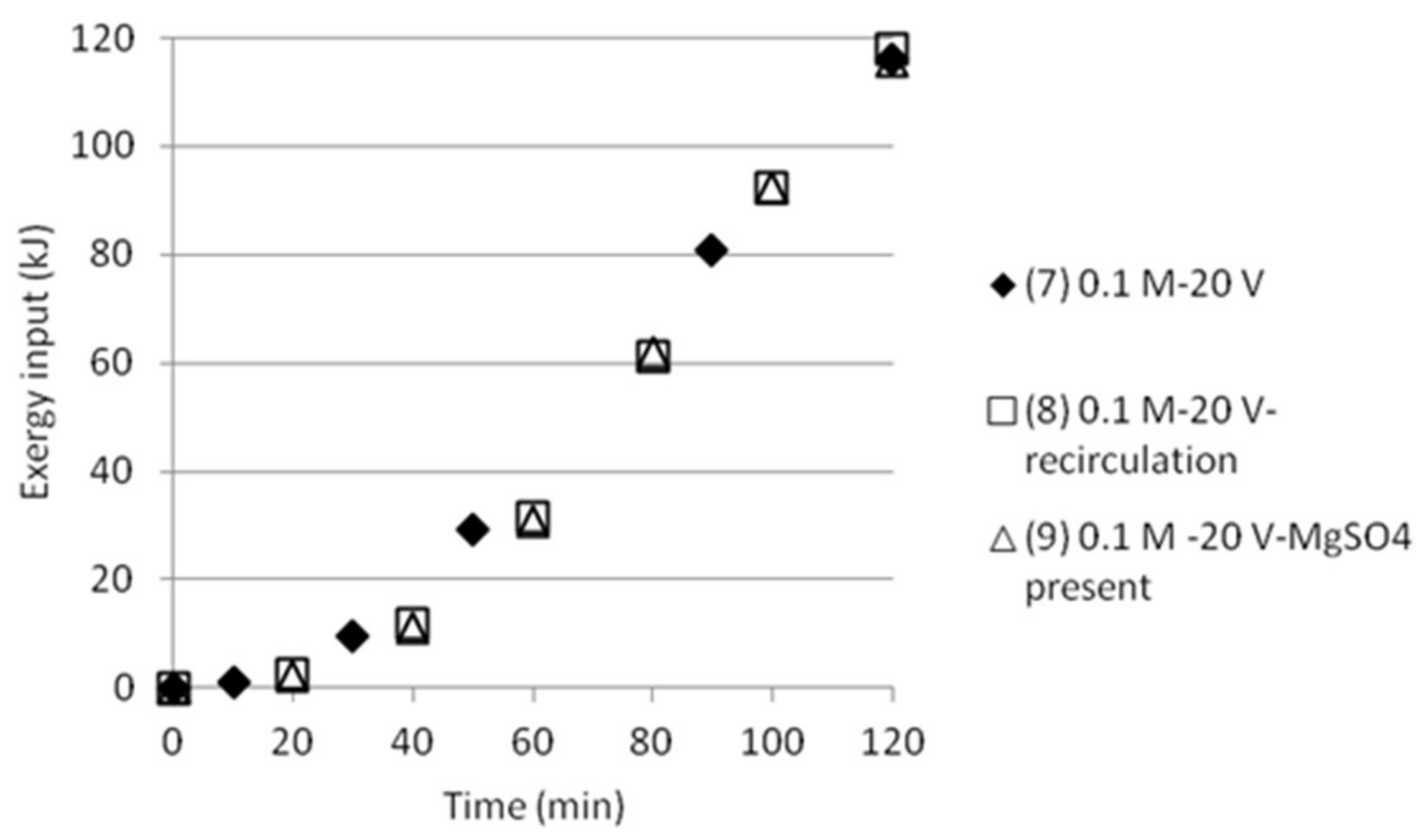
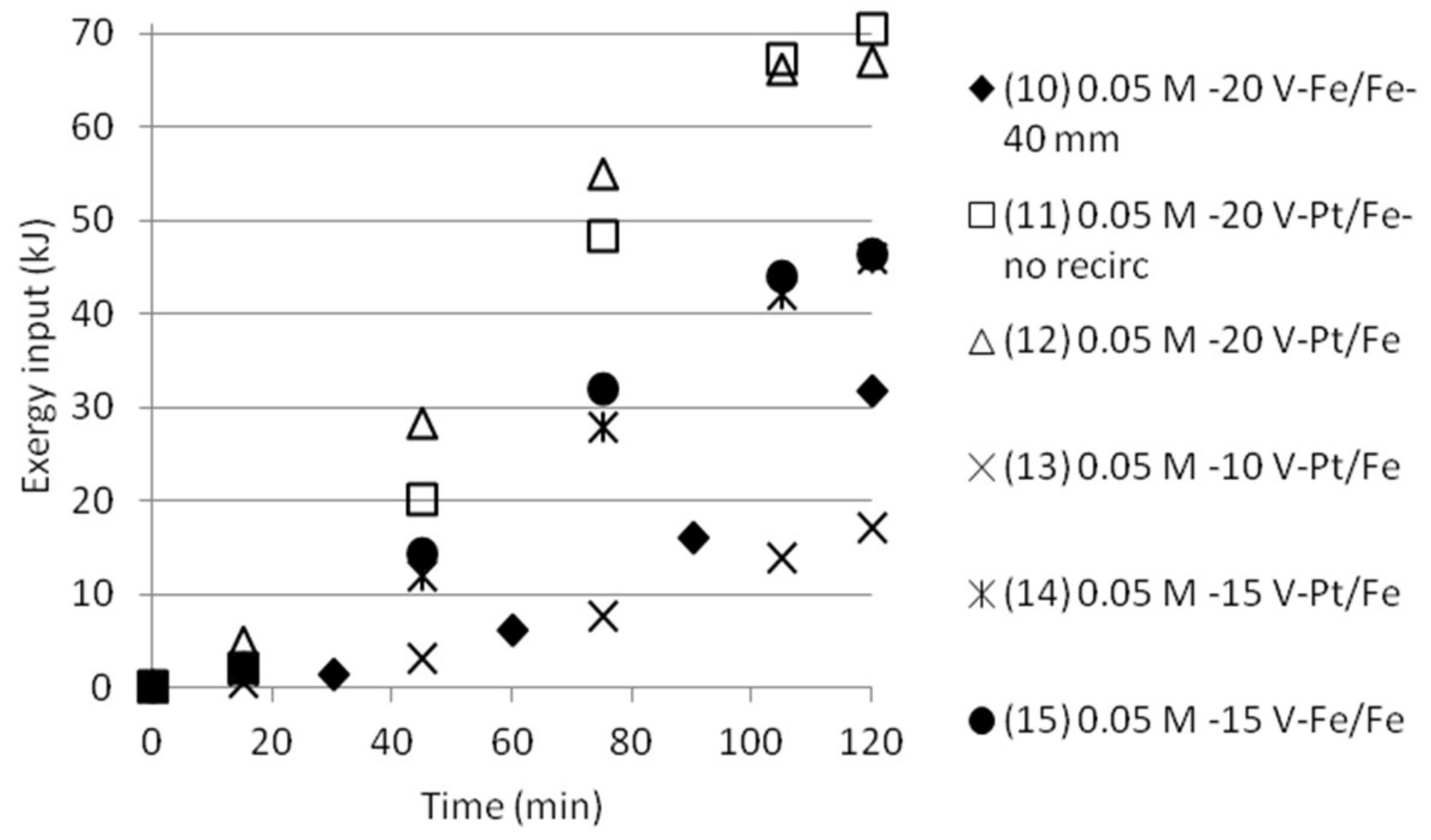
3.5. Exergy Calculations
3.5.1. Three Compartments
3.5.2. Double Cell Distance and Five Compartments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABS | ammonium bisulfate, NH4HSO4 | |
| ACS | monovalent anion exchange membrane | |
| AS | ammonium sulfate, (NH4)2SO4 | |
| CMS | monovalent cation exchange membrane | |
| DC | direct current | |
| ISE | ion selective electrode | |
| RO | reverse osmosis | |
| Symbols | ||
| Exin,mix | input exergy, exergy of mixing | J (kJ, kJ/NH4+) |
| I | electrical current | A (mA) |
| n ip,if | molar amount of I in product, feed stream | mole |
| Ka | equilibrium constant | - |
| pKa | logarithmic equilibrium constant | - |
| P | power | W |
| R | gas constant (8.31451) | J/(mol·K) |
| T | temperature | °C |
| t | time | s |
| U | voltage | V |
| xi | molar fraction of i | mole/mole |
| γi | activity coefficient for dissolved species i | - |
| Δ | difference | |
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane | CMS | ACS |
|---|---|---|
| Type | Strong Acid (Na type) | Strong Base (Cl type) |
| Characteristics | Monovalent cation permselectivity | Monovalent anion permselectivity |
| Electric resistance | 1.8 Ω·cm2 | 3.8 Ω·cm2 |
| Burst strength | >0.15 MPa | >0.15 MPa |
| Thickness | 0.15 mm | 0.13 mm |
| Temperature | <40 °C | <40 °C |
| pH | 0–10 | 0–8 |
Appendix C
Appendix D

References
- Nduagu, E. Production of Mg(OH)2 from Mg-Silicate Rock for CO2 Sequestration. Ph.D. Thesis, Åbo Akademi University, Turku, Finland, 2012. Available online: http://urn.fi/URN:NBN:fi-fe201311117327. Available online: http://urn.fi/URN:NBN:fi-fe201311117327 (accessed on 24 February 2018).
- Romão, I.S. Production of Magnesium Carbonates from Serpentinites for CO2 Mineral Sequestration—Optimisation towards Industrial Application. Ph.D. Thesis, Åbo Akademi University/University of Coimbra, Turku/Coimbra, Finland/Portugal, 2015. [Google Scholar]
- Virtanen, M. Energy-efficient solutions for concentrating or separating dissolved ammonium sulphate. M.Sc. Thesis, Thermal and Flow Engineering Laboratory, Åbo Akademi University, Turku, Finland, 2015. [Google Scholar]
- Tanaka, Y. Ion Exchange Membrane Electrodialysis; Nova Science Publishers, Inc.: New York, NY, USA, 2010. [Google Scholar]
- Erlund, R.; Koivisto, E.; Fagerholm, M.; Zevenhoven, R. Extraction of magnesium from four Finnish magnesium silicate rocks for CO2 mineralisation—Part 2: Aqueous solution extraction. Hydrometallurgy 2016, 166, 229–236. [Google Scholar] [CrossRef]
- Koivisto, E.; Zevenhoven, R. Methods for recovery and re-use of additive chemicals during CO2 mineralisation. J. Water Proc. Eng. 2017, 20, 61–70. [Google Scholar] [CrossRef]
- Zevenhoven, R.; Slotte, M.; Åbacka, J.; Highfield, J. A comparison of CO2 mineral carbonation processes involving a dry or wet carbonation step. Energy 2016, 117, 604–611. [Google Scholar] [CrossRef]
- Campione, A.; Gurreri, L.; Ciofalo, M.; Micale, G.; Tamburini, A.; Cipollina, A. Electrodialysis for water desalination: A critical assessment of recent developments on process fundamentals, models and applications. Desalination 2018, 434, 121–160. [Google Scholar] [CrossRef]
- Yusuf, I.D.Y.; Ompusunggu, S.R.P.; Bagastyo, A.Y. Salt recovery from reverse osmosis concentrate using electrodialysis. In Proceedings of the 5th Environmental Technology and Management Conference, Bandung, Indonesia, 23–24 November 2015. [Google Scholar]
- Van der Bruggen, B.; Koninckx, A.; Vandecasteele, C. Separation of monovalent and divalent ions from aqueous solution by electrodialysis and nanofiltration. Water Res. 2004, 38, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Koter, S.; Chojnowska, P.; Szynkiewicz, K.; Koter, I. Batch electrodialysis of ammonium nitrate and sulfate solutions. J. Membr. Sci. 2015, 496, 219–228. [Google Scholar] [CrossRef]
- Gao, L.; Liu, D.; Yang, J. Modeling of a three-compartment membrane electrodialysis in H2SO4–(NH4)2SO4–NH3·H2O system. J. Membr. Sci. 2009, 344, 252–257. [Google Scholar] [CrossRef]
- Galama, A.H.; Daubares, G.; Burheim, O.S.; Rijnaarts, H.H.M.; Post, J.W. Seawater electrodialysis with preferential removal of divalent ions. J. Membr. Sci. 2014, 452, 219–228. [Google Scholar] [CrossRef]
- Xu, X.; He, Q.; Ma, G.; Wang, H.; Nirmalakhandan, N.; Xu, P. Selective separation of mono- and di-valent cations in electrodialysis during brackish water desalination: Bench and pilot-scale studies. Desalination 2018, 428, 146–160. [Google Scholar] [CrossRef]
- Szargut, J.; Morris, D.; Steward, F.R. Exergy analysis of thermal, chemical and metallurgical processes; Hemisphere Publishing Co.: New York, NY, USA, 1989. [Google Scholar]
- Clegg, S.L.; Milioto, S.; Palmer, D.A. Osmotic and Activity Coefficients of Aqueous (NH4)2SO4 as a Function of Temperature, and Aqueous (NH4)2SO4-H2SO4 Mixtures at 298.15 K and 323.15 K. J. Chem. Eng. Data 1996, 41, 455–467. [Google Scholar] [CrossRef]
- Noble, R.D.; Stern, S.A. Membrane Separations Technology: Principles and Applications; Elsevier: Amsterdam, Netherlands, 1995; p. 249. [Google Scholar]



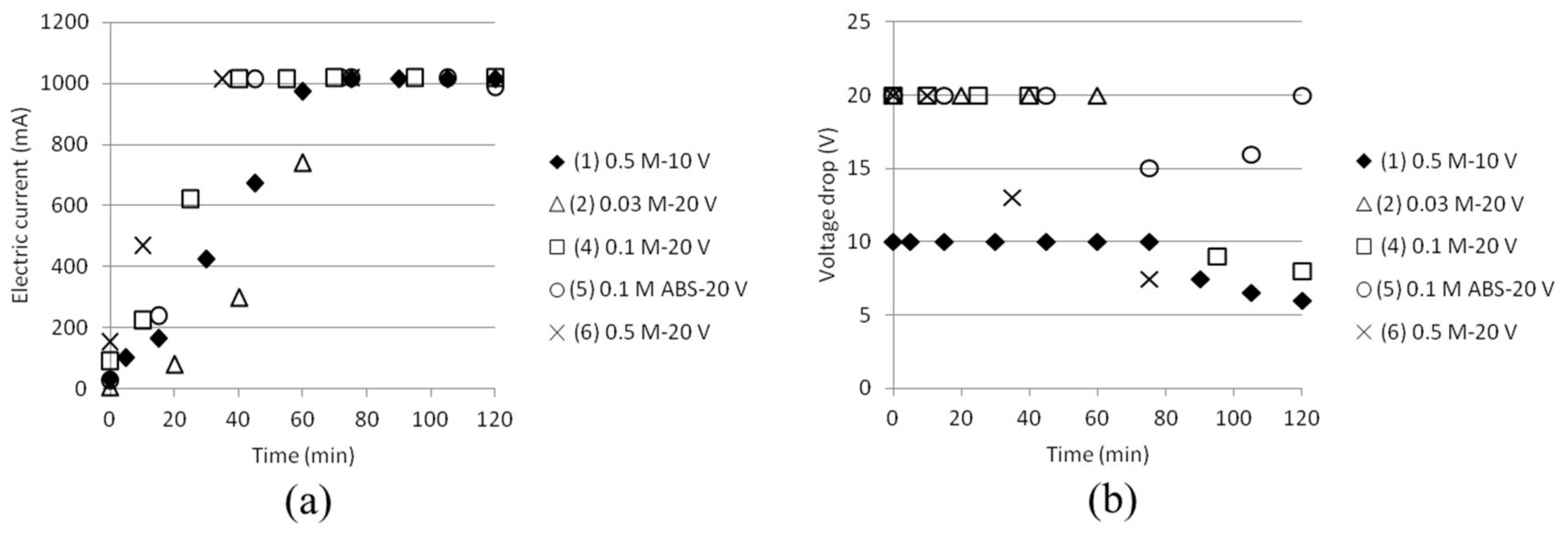
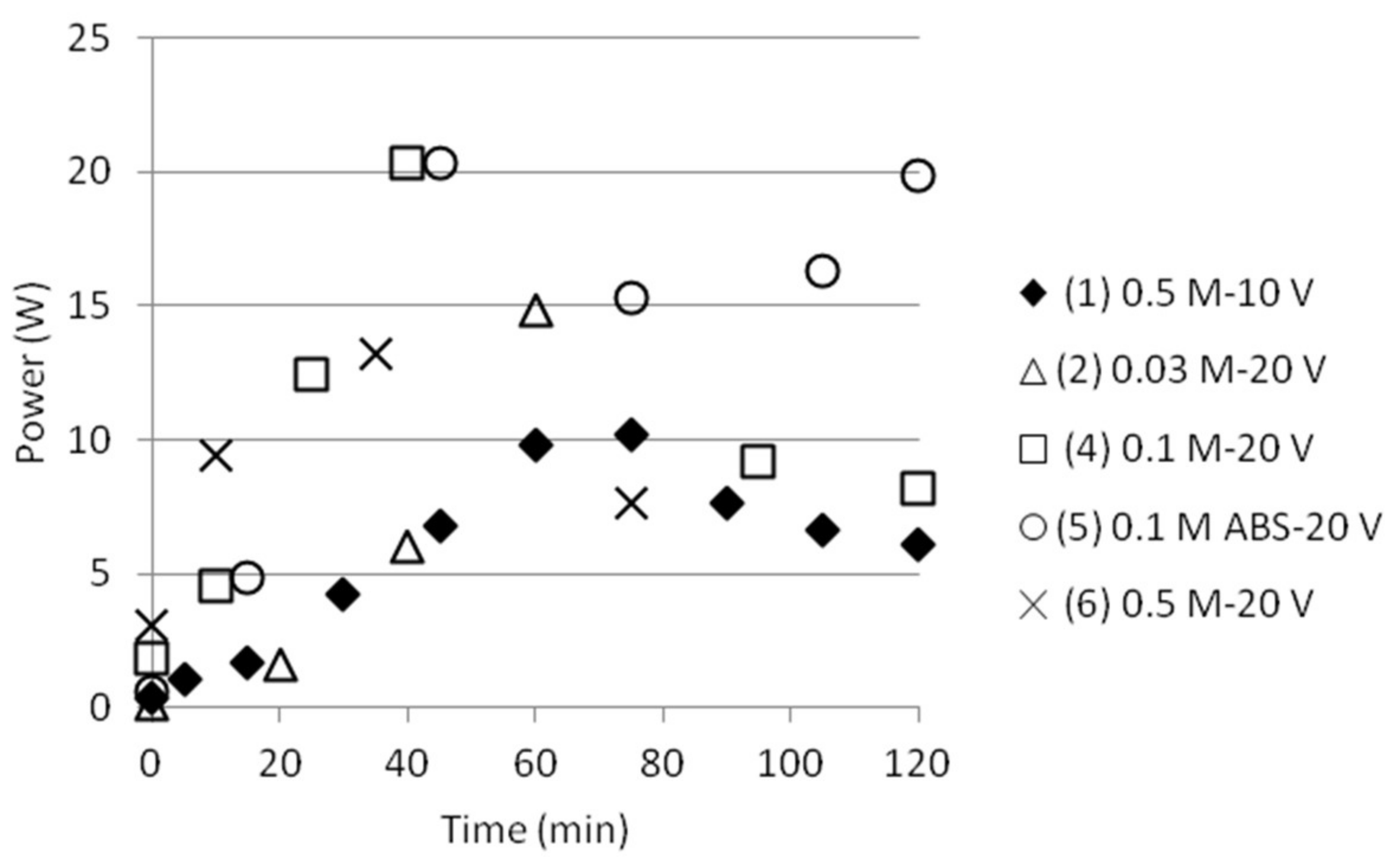
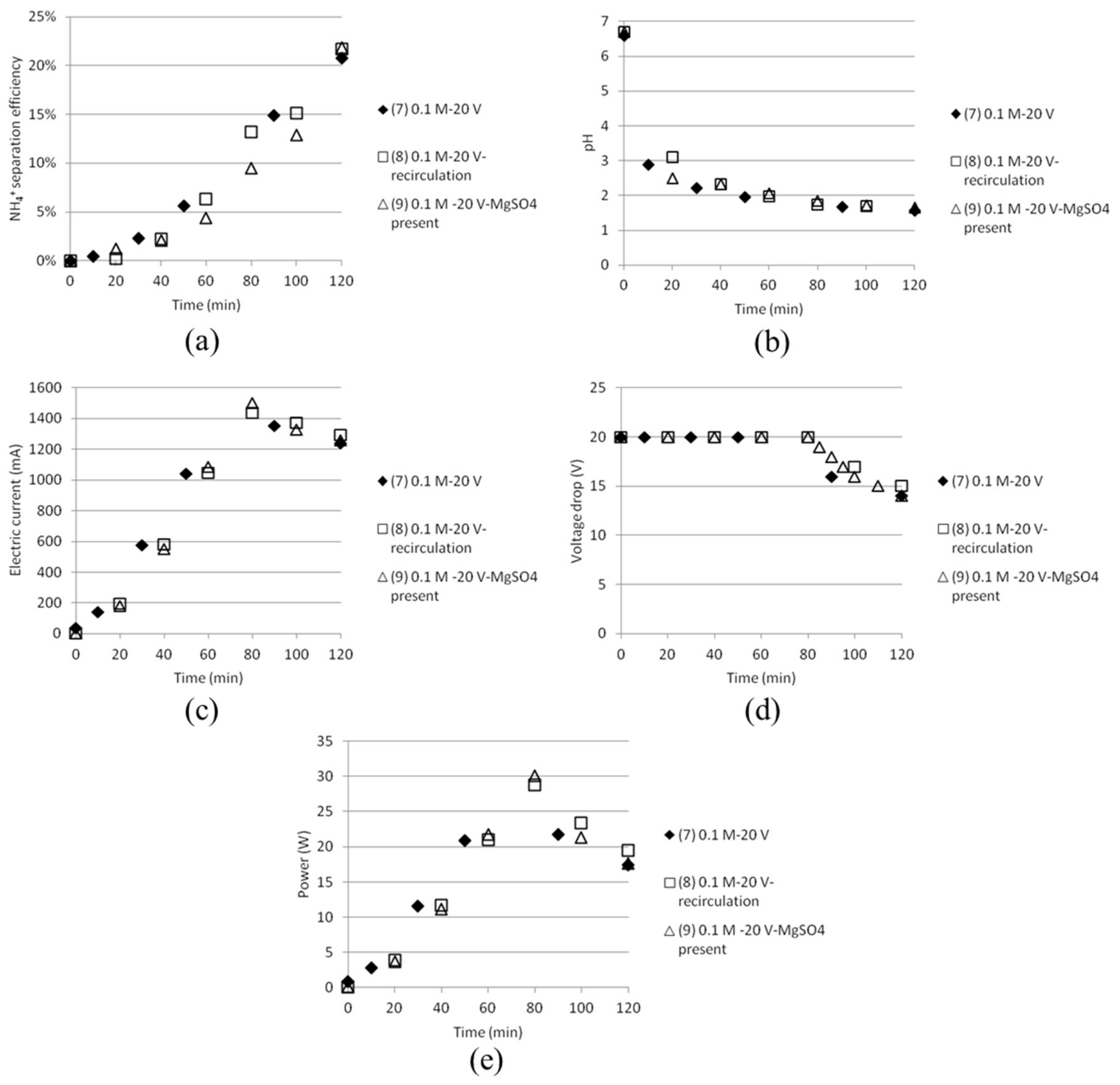
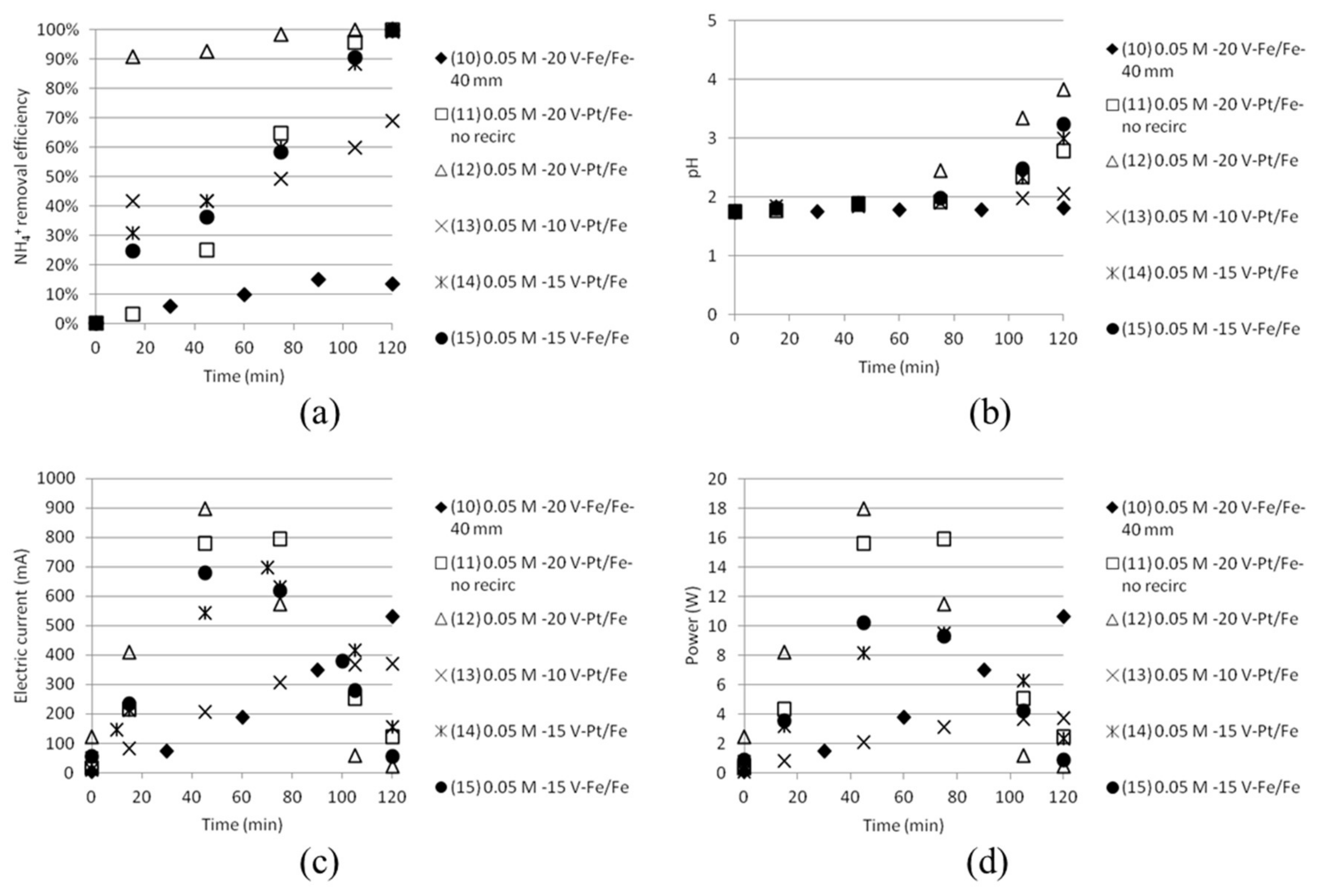
| Solution | Time (h) | AS/ABS (Cell 1) | Dest. H2O (Cell 2) | AS/ABS (Cell 3) |
|---|---|---|---|---|
| pH | 0 | 1.92 | 6.7 | 1.92 |
| pH | 24 | 1.83 | 2.45 | 2.75 |
| pH | 48 | 1.82 | 2.35 | 2.52 |
| Conductivity, μS/cm | 0 | 14450 | 10 | 14450 |
| Conductivity, μS/cm | 24 | 13100 | 2100 | 9800 |
| Conductivity, μS/cm | 48 | 14700 | 2500 | 10100 |
| [NH4+], mmol/dm3 | 0 | 49.7 | 0.05 | 49.7 |
| [NH4+], mmol/dm3 | 24 | No data | No data | No data |
| [NH4+], mmol/dm3 | 48 | 41 | 6 | 59 |
| [NH4+] balance, % | 48 | 8.7↓ | 5.95↑ | 9.3↑ |
| Test | Time (min) | Anode/Cathode | Voltage (V) | AS (mol/dm3) | ABS (mol/dm3) |
|---|---|---|---|---|---|
| 1 | 120 | Stainless steel (×2) | 10 | 0.5 | 0.5 |
| 2 | 60 | Stainless steel (×2) | 20 | 0.03 | 0.03 |
| 3 | 25 | Silver/Stainless steel | 20 | 0.03 | 0.03 |
| 4 | 120 | Stainless steel (×2) | 20 | 0.1 | 0.1 |
| 5 | 120 | Stainless steel (×2) | 20 | - | 0.1 |
| 6 | 75 | Stainless steel (×2) | 20 | 0.5 | 0.5 |
| Test | Test Name | Time (min) | pHfinal | [NH4+]start as ABS | [NH4+]final | SO42− /HSO4−final | [HSO4−]final | ABS sep. eff. (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.5 M-10 V | 120 | 1.75 | 0.268 | 0.072 | 0.575 | 0.0559 | 20.9 |
| 2 | 0.03 M-20 V | 60 | 1.99 | 0.017 | 0.013 | 1.000 | 0.0087 | 50.7 |
| 4 | 0.1 M-20 V | 120 | 1.48 | 0.057 | 0.058 | 0.309 | 0.0502 | 88.1 |
| 5 | 0.1 M ABS-20 V | 120 | 1.43 | 0.057 | 0.021 | 0.275 | 0.0180 | 31.6 |
| 6 | 0.5 M-20 V | 75 | 1.94 | 0.268 | 0.072 | 0.891 | 0.0498 | 18.6 |
| Test | Time (min) | Anode/Cathode | Voltage (V) | AS (mol/dm3) | ABS (mol/dm3) | MgSO4 (mol/dm3) | Recirculation (20 mL/min) |
|---|---|---|---|---|---|---|---|
| 7 | 120 | Stainless steel (×2) | 20 | 0.1 | 0.1 | - | No |
| 8 | 120 | Stainless steel (×2) | 20 | 0.1 | 0.1 | - | Yes |
| 9 | 120 | Stainless steel (×2) | 20 | 0.1 | 0.1 | 0.1 | Yes |
| Test | Time (min) | Anode/Cathode | Voltage (V) | AS/ABS (mol/dm3) | Cell Width (mm) | Recirculation (20 mL/min) |
|---|---|---|---|---|---|---|
| 10 | 120 | Stainless steel (×2) | 20 | 0.05/0.05 | 40 | Yes |
| 11 | 120 | Platinum/Stainless steel | 20 | 0.05/0.05 | 18 | No |
| 12 | 120 | Platinum/Stainless steel | 20 | 0.05/0.05 | 18 | Yes |
| 13 | 120 | Platinum/Stainless steel | 10 | 0.05/0.05 | 18 | Yes |
| 14 | 120 | Platinum/Stainless steel | 15 | 0.05/0.05 | 18 | Yes |
| 15 | 120 | Stainless steel (×2) | 15 | 0.05/0.05 | 18 | Yes |
| Test | Test Name | Cell 1 ∆Exmix (kJ) | Cell 1 ∆Exmix (kJ/mol NH4+) | Cell 2 ∆Exmix (kJ) | Cell 2 ∆Exmix (kJ/mol NH4+) |
|---|---|---|---|---|---|
| 1 | 0.5 M-10 V | 0.0365 | 0.0093 | −0.0030 | −0.0212 |
| 2 | 0.03 M-20 V | 0.0008 | 0.0033 | −0.0002 | −.0048 |
| 4 | 0.1 M-20 V | 0.0009 | 0.0063 | −0.0024 | −0.0176 |
| 5 | 0.1 M ABS-20 V | - | - | −0.0007 | −0.0072 |
| 6 | 0.5 M-20 V | - | - | −0.0030 | −0.0212 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koivisto, E.; Zevenhoven, R. Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation. Geosciences 2018, 8, 123. https://doi.org/10.3390/geosciences8040123
Koivisto E, Zevenhoven R. Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation. Geosciences. 2018; 8(4):123. https://doi.org/10.3390/geosciences8040123
Chicago/Turabian StyleKoivisto, Evelina, and Ron Zevenhoven. 2018. "Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation" Geosciences 8, no. 4: 123. https://doi.org/10.3390/geosciences8040123
APA StyleKoivisto, E., & Zevenhoven, R. (2018). Membrane Separation of Ammonium Bisulfate from Ammonium Sulfate in Aqueous Solutions for CO2 Mineralisation. Geosciences, 8(4), 123. https://doi.org/10.3390/geosciences8040123