
Comparative Mitogenomic Analyses of Tanypodinae (Diptera: Chironomidae)
 , , , , ,
, , , , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and Specimen Identification
2.2. Sequencing, Assembly, and Annotation
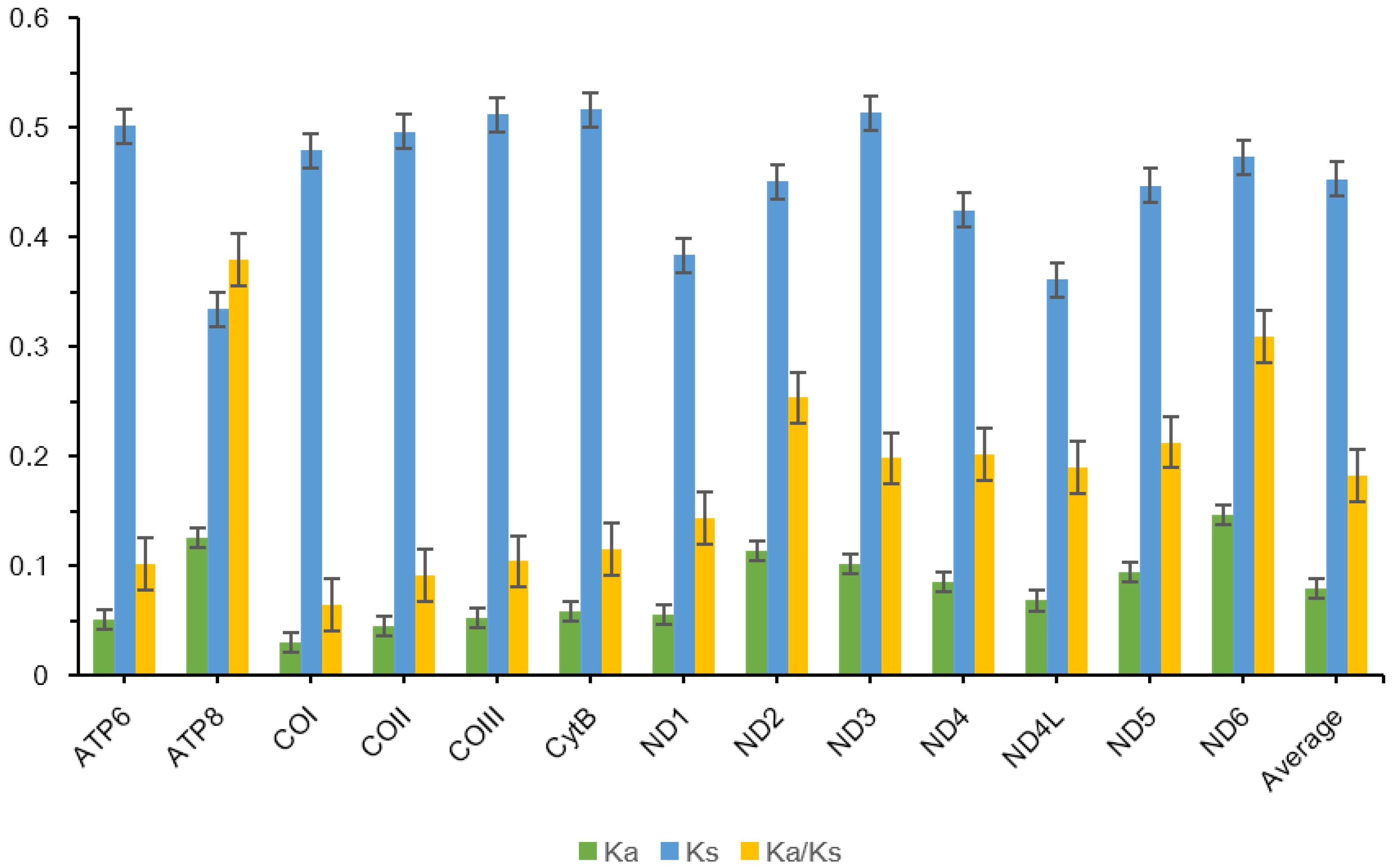
2.3. Sequence Analysis and Substitution Rate
2.4. Phylogenetic Analyses
3. Results and Discussions
3.1. General Features of 21 Tanypodinae and 1 Podonominae Mitogenomes
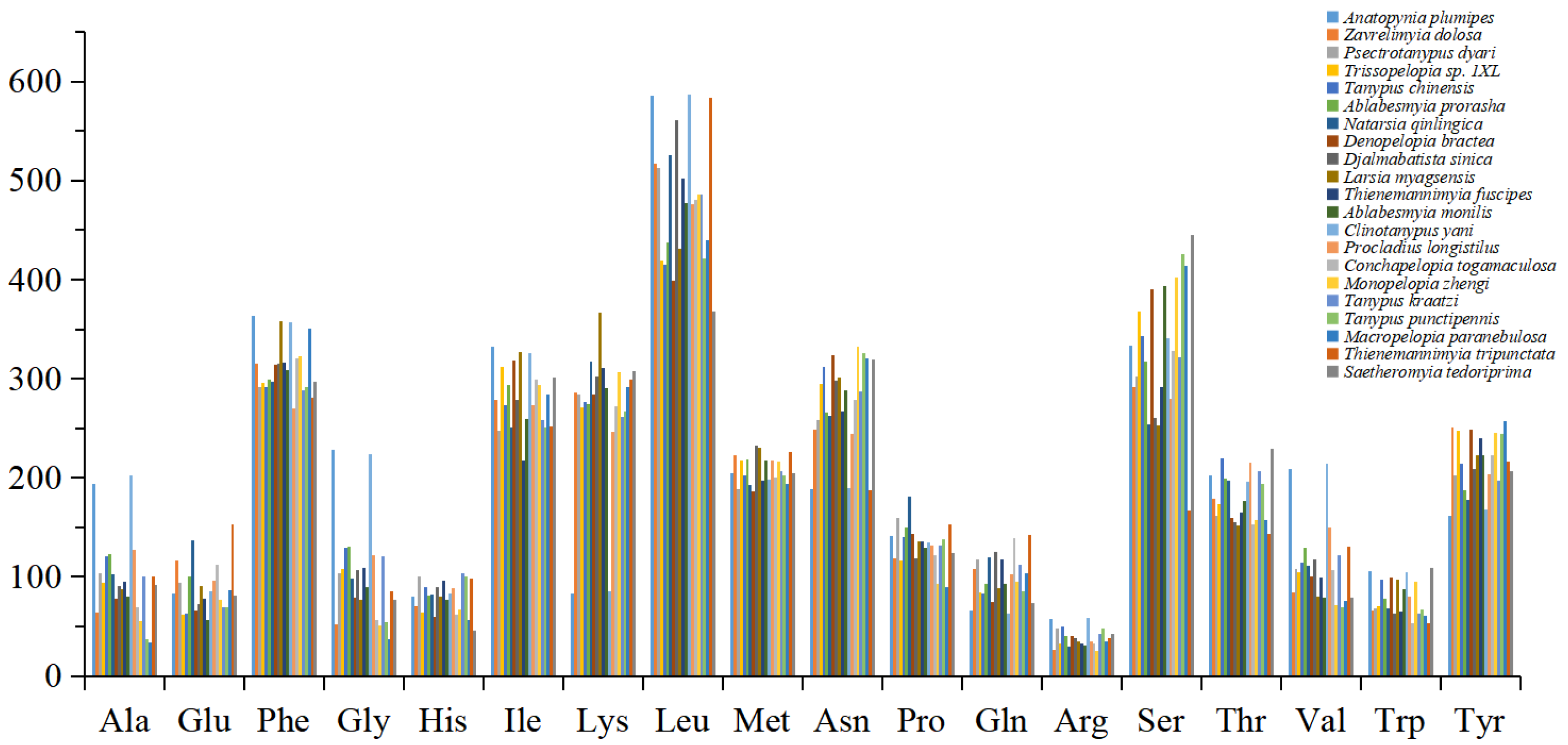
3.2. Protein-Coding Genes
3.3. Mitochondrial Gene Codon Usage
3.4. Transfer and Ribosomal RNA Genes
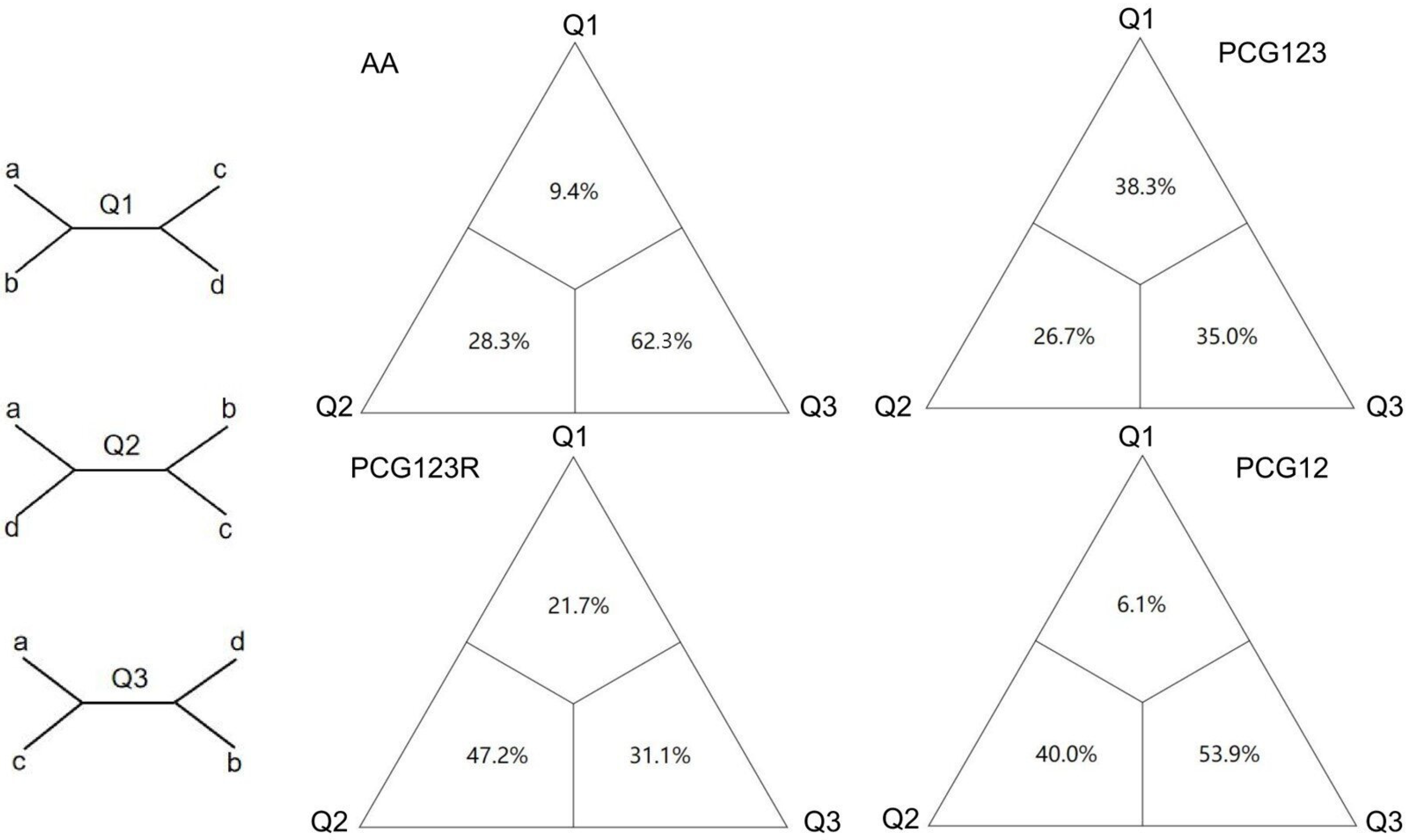
3.5. Heterogeneity Analysis
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.-Y.; Yan, L.-P.; Pape, T.; Gao, Y.-Y.; Zhang, D. Evolutionary insights into bot flies (Insecta: Diptera: Oestridae) from comparative analysis of the mitochondrial genomes. Int. J. Biol. Macromol. 2020, 149, 371–380. [Google Scholar] [CrossRef]
- Li, S.-Y.; Zhao, Y.-M.; Guo, B.-X.; Li, C.-H.; Sun, B.-J.; Lin, X.-L. Comparative analysis of mitogenomes of Chironomus (Diptera: Chironomidae). Insects 2022, 13, 1164. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.L.D.; Ekrem, T. Phylogenetic relationships of nonbiting midges in the subfamily Tanypodinae (Diptera: Chironomidae) inferred from morphology. Syst. Entomol. 2016, 41, 73–92. [Google Scholar] [CrossRef]
- Zheng, C.-G.; Liu, Z.; Zhao, Y.-M.; Wang, Y.; Bu, W.-J.; Wang, X.-H.; Lin, X.-L. First report on mitochondrial gene rearrangement in non-biting midges, revealing a synapomorphy in Stenochironomus Kieffer (Diptera: Chironomidae). Insects 2022, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, F.-X.; Li, X.-B.; Aishan, Z.; Lin, X.-L. New mitogenomes of the Polypedilum generic complex (Diptera: Chironomidae): Characterization and phylogenetic implications. Insects 2023, 14, 238. [Google Scholar] [CrossRef]
- Allegrucci, G.; Carchini, G.; Todisco, V.; Convey, P.; Sbordoni, V. A molecular phylogeny of Antarctic Chironomidae and its implications for biogeographical history. Polar Biol. 2006, 29, 320–326. [Google Scholar] [CrossRef]
- Cranston, P.S.; Hardy, N.B.; Morse, G.E. A dated molecular phylogeny for the Chironomidae (Diptera). Syst. Entomol. 2012, 37, 172–188. [Google Scholar] [CrossRef]
- Siri, A.; Donato, M. Phylogenetic analysis of the tribe Macropelopiini (Chironomidae: Tanypodinae): Adjusting homoplasies. Zool. J. Linn. Soc. 2015, 174, 74–92. [Google Scholar] [CrossRef]
- Zheng, C.-G.; Zhu, X.-X.; Yan, L.-P.; Yao, Y.; Bu, W.-J.; Wang, X.-H.; Lin, X.-L. First complete mitogenomes of Diamesinae, Orthocladiinae, Prodiamesinae, Tanypodinae (Diptera: Chironomidae) and their implication in phylogenetics. PeerJ 2021, 9, e11294. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Zheng, X.; Song, C.; Jin, H.; Chen, L.; Qi, X. Limited Variation in Codon Usage across Mitochondrial Genomes of Non-Biting Midges (Diptera: Chironomidae). Insects 2024, 15, 752. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, C.; Tang, Y.; Zhang, Y.; Ge, X.; Zhang, J.; Liu, W. Complete Mitochondrial Genome of Tanypus chinensis and Tanypus kraatzi (Diptera: Chironomidae): Characterization and Phylogenetic Implications. Genes 2024, 15, 1281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-W.; Zhao, Y.-M.; Lin, X.-L. First report of the complete mitogenome of Tanypus punctipennis Meigen, 1818 (Diptera, Chironomidae) from Hebei Province, China. Mitochondrial DNA B 2022, 7, 215–216. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Manchekar, M.; Scissum-Gunn, K.; Song, D.; Khazi, F.; Mclean, S.L.; Nielsen, B.L. DNA recombination activity in soybean mitochondria. J. Mol. Biol. 2006, 356, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, X.; Yin, X.; Li, X.; Xi, Y. The mitochondrial genomes of ladybird beetles and implications for evolution and phylogeny. Int. J. Biol. Macromol. 2020, 147, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wang, J.; Zang, H.; Chai, L.; Liu, W.; Zhang, J.; Yan, C.; Wang, B. Mitogenomics Provide New Phylogenetic Insights of the Family Apataniidae (Trichoptera: Integripalpia). Insects 2024, 15, 973. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Peng, L.; Vogler, A.P.; Morse, J.C.; Yang, L.; Sun, C.; Wang, B. Massive gene rearrangements of mitochondrial genomes and implications for the phylogeny of Trichoptera (Insecta). Syst. Entomol. 2023, 48, 278–295. [Google Scholar] [CrossRef]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Bian, D.; Dai, M.; Ye, W.; Lu, Z.; Li, M.; Fang, Y.; Qu, J.; Su, W.; Li, F.; Sun, H. Complete mitochondrial genome of Spilosoma lubricipedum (Noctuoidea: Erebidae) and implications for phylogeny of noctuid insects. Genomics 2020, 112, 4577–4584. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Liu, J.; Chiba, H.; Xiao, J.; Yuan, X. Complete mitochondrial genomes of three skippers in the tribe Aeromachini (Lepidoptera: Hesperiidae: Hesperiinae) and their phylogenetic implications. Ecol. Evol. 2021, 11, 8381–8393. [Google Scholar] [CrossRef]
- Ge, X.; Peng, L.; Morse, J.C.; Wang, J.; Zang, H.; Yang, L.; Sun, C.; Wang, B. Phylogenomics resolves a 100-year-old debate regarding the evolutionary history of caddisflies (Insecta: Trichoptera). Mol. Phylogenet. Evol. 2024, 201, 108196. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, X. Denopelopia Roback & Rutter from China with emendation of the generic diagnosis (Diptera: Chironomidae: Tanypodinae). Zootaxa 2005, 1042, 55–63. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, X.H. Natarsia Fittkau (Diptera: Chironomidae: Tanypodinae) from China. Zootaxa 2006, 1111, 59–67. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, X. New species of Clinotanypus Kieffer, 1913 (Chironomidae: Tanypodinae) from China. Zootaxa 2008, 1944, 53–65. [Google Scholar] [CrossRef]
- Duan, X.; Chang, T.; Jiao, K.-L.; Wang, X.-H.; Lin, X.-L. Monopelopia Fittkau, 1962, a newly recorded genus from Oriental China (Diptera, Chironomidae) with a description of Monopelopia zhengi Lin sp. n. Zootaxa 2021, 4980, 383388. [Google Scholar] [CrossRef]
- Liu, J.; Tang, H. Djalmabatista Fittkau, 1968 (Diptera: Chironomidae: Tanypodinae) from Oriental China, with the description of a new species. Pan-Pac. Entomol. 2017, 93, 14–21. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Shin, S.C. Complete mitochondrial genome of the Antarctic midge Parochlus steinenii (Diptera: Chironomidae). Mitochondrial DNA A 2016, 27, 3475–3476. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE–visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.A.; Von Haeseler, A. Maximum-likelihood analysis using TREE-PUZZLE. Curr. Protoc. Bioinform. 2007, 17, 6.6.1–6.6.23. [Google Scholar] [CrossRef] [PubMed]
- Oteo-Garcia, G.; Oteo, J.A. Evolutionary distances corrected for purifying selection and ancestral polymorphisms. J. Theor. Biol. 2019, 483, 110004. [Google Scholar] [CrossRef] [PubMed]
- Johri, P.; Charlesworth, B.; Jensen, J.D. Toward an evolutionarily appropriate null model: Jointly inferring demography and purifying selection. Genetics 2020, 215, 173–192. [Google Scholar] [CrossRef]
- James, J.E.; Piganeau, G.; Eyre-Walker, A. The rate of adaptive evolution in animal mitochondria. Mol. Ecol. 2016, 25, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, H.; Zhao, T. Insights into the phylogeny of Hemiptera from increased mitogenomic taxon sampling. Mol. Phylogenet. Evol. 2019, 137, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Krosch, M.; Cranston, P.; Bryant, L.; Strutt, F.; Mccluen, S. Towards a dated molecular phylogeny of the Tanypodinae (Chironomidae, Diptera). Invertebr. Syst. 2017, 31, 302–316. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Sample ID | Life Stage | Sampling Metadata | GenBank Accession | Reference |
|---|---|---|---|---|---|---|
| Tanypodinae | Ablabesmyia monilis | XL1622 | Adult male | Antu, Yanbian, Jilin, China, 42.457° N, 128.144° E, 12 July 2016, leg. C. Song. | OP006242 | this study |
| Tanypodinae | Ablabesmyia prorasha | sun319 | Adult male | Baisha, Hainan, China, 19.116° N, 109.092° E, leg. Y. Fu. | OP006228 | this study |
| Tanypodinae | Anatopynia plumipes | CHLA111 | Larva | Zhalong wetland, Heilongjiang, China, 47.168° N, 124.172° E, 1 October 2019, leg. M.-H. Liu | OP006225 | this study |
| Tanypodinae | Clinotanypus yani | XL2042 | Adult male | Jiulongshan Nature Reserve, Guangyuan, Sichuan, China, 31.976° N, 106.036° E, 8 August 2017, leg. C. Song | MW373524 | [9] |
| Tanypodinae | Conchapelopia togamaculosa | XL2451 | Adult male | Xuanen, Enshi, Hubei, China, 29.669° N, 109.609° E, 11 July 2015, leg. B.-J. Sun | OP006233 | this study |
| Tanypodinae | Denopelopia bractea | sun359 | Adult male | Kaihua, Quzhou, Zhejiang, China, 29.177° N, 118.120° E, 17 April 2011, leg. X.-L. Lin | OP006240 | this study |
| Tanypodinae | Djalmabatista sinica | sun456 | Adult male | Changjiang, Hainan, China, 19.116° N, 109.092° E, 31 March 2016, leg. B.-J. Sun | OP006241 | this study |
| Tanypodinae | Larsia myagsensis | sun465 | Adult male | Yinggeling, Baisha, Hainan, China, 19.083° N, 109.500° E, 14 March 2016, leg. B.-J. Sun | OP006230 | this study |
| Tanypodinae | Macropelopiaparanebulosa | XL2893 | Larva | Benxi, Liaoning, China, 41.289° N, 124.898° E, 3 September 2014, leg. C. Song | OP006236 | this study |
| Tanypodinae | Monopelopia zhengi | XL2560 | Adult male | Ledong, Hainan, China, 18.693° N, 108.796° E, 8 March 2016, leg. B.-J. Sun | OP006234 | this study |
| Tanypodinae | Natarsiaqinlingica | sun342 | Adult male | Yinzhou, Ningbo, Zhejiang, China, 29.804° N, 121.788° E, 10 May 2012, leg. X.-L. Lin | OP006229 | this study |
| Tanypodinae | Psectrotanypus dyari | NLCH409 | Larva | Shixing, Shaoguan, Guangdong, China, 24.723° N, 114.257° E, 24 August 2020, leg. X.-L. Lin | OP006244 | this study |
| Tanypodinae | Procladius longistilus | XL2158 | Adule female | Luotian, Huanggang, Hubei, China, 31.099° N, 115.734° E, 7 July 2019, leg. S. Qiu | OP006232 | this study |
| Tanypodinae | Saetheromyia tedoriprima | XL3064 | Adult male | Menghai, Xishuangbanna, Yunnan, China, 22.055° N, 99.990° E, 6 May 2013, leg. X.-L. Lin | OP006243 | this study |
| Tanypodinae | Tanypuschinensis | sun245 | Adult male | Deqing, Huzhou, Zhejiang, China, 28.980° N, 118.959° E, leg. 15 July 2012, leg. X.-L. Lin | OP006227 | this study |
| Tanypodinae | Tanypus kraatzi | XL2603 | Larva | Baoding, Hebei, China, 38.321° N, 115.375° E, 9 May 2018, leg. X.-L. Lin | OP006235 | this study |
| Tanypodinae | Tanypus punctipennis | XL2604 | Larva | Baoding, Hebei, China, 38.320° N, 115.375° E, 9 May 2018, leg. X.-L. Lin | MZ475054 | [12] |
| Tanypodinae | Thienemannimyia fuscipes | XL1540 | Adult male | Wuying, Yichun, Heilongjiang, China, 48.087° N, 129.247° E, 27 July 2016, leg. C. Song | OP006231 | this study |
| Tanypodinae | Thienemannimyia tripunctata | XL3034 | Adult male | Mengla, Xishuangbanna, Yunnan, China, 21.928° N, 101.255° E, 22 April 2014, leg. Q. Wang | OP006237 | this study |
| Tanypodinae | Trissopelopia sp. 1XL | NLCH909 | Larva | Long, Guilin, Guangxi, China, 25.625° N, 109.914° E, 16 August 2020, leg. Y. Yao | OP006226 | this study |
| Tanypodinae | Zavrelimyia dolosa | LJHZ33 | Adult male | Hechi, Guangxi, China, 24.731° N, 107.896° E, 5 June 2020, leg. Z.-N. Yang | OP006239 | this study |
| Podonominae | Paraboreochlus okinawanus | LGS290 | Adult male | Leishan, Qiandongnan, Guizhou, China, 26.396° N, 108.260° E, 18 January 2021, leg. H.-J. Yu | OP006238 | this study |
| Podonominae | Parochlus steinenii | KT003702 | King George Island, West, Antarctica, 62.233° S, 58.783° W, summer in 2015 | KT003702 | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, X.-R.; Chen, M.-H.; Li, S.-Y.; Guo, B.-X.; Zhang, Y.; Zhang, Z.-C.; Qiao, Y.-J.; Lin, X.-L. Comparative Mitogenomic Analyses of Tanypodinae (Diptera: Chironomidae). Insects 2025, 16, 203. https://doi.org/10.3390/insects16020203
Xiao X-R, Chen M-H, Li S-Y, Guo B-X, Zhang Y, Zhang Z-C, Qiao Y-J, Lin X-L. Comparative Mitogenomic Analyses of Tanypodinae (Diptera: Chironomidae). Insects. 2025; 16(2):203. https://doi.org/10.3390/insects16020203
Chicago/Turabian StyleXiao, Xiu-Ru, Meng-Han Chen, Shu-Yi Li, Bing-Xin Guo, Yan Zhang, Zhi-Chao Zhang, Ya-Jun Qiao, and Xiao-Long Lin. 2025. "Comparative Mitogenomic Analyses of Tanypodinae (Diptera: Chironomidae)" Insects 16, no. 2: 203. https://doi.org/10.3390/insects16020203
APA StyleXiao, X.-R., Chen, M.-H., Li, S.-Y., Guo, B.-X., Zhang, Y., Zhang, Z.-C., Qiao, Y.-J., & Lin, X.-L. (2025). Comparative Mitogenomic Analyses of Tanypodinae (Diptera: Chironomidae). Insects, 16(2), 203. https://doi.org/10.3390/insects16020203