
Microbiome of Invasive Tick Species Haemaphysalis longicornis in North Carolina, USA

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tick Collection and Identification
2.2. DNA Extraction and Library Construction
2.3. Bacterial 16S rRNA Gene Amplification and Library Preparation
2.4. Bioinformatics Data Processing and Statistical Analyses
2.5. Phylogenetic Analyses of Aeromonas, Coxiella, Rickettsia, and Staphylococcus Sequences
3. Results
3.1. 16S rRNA V3-V4 Region Sequencing Results
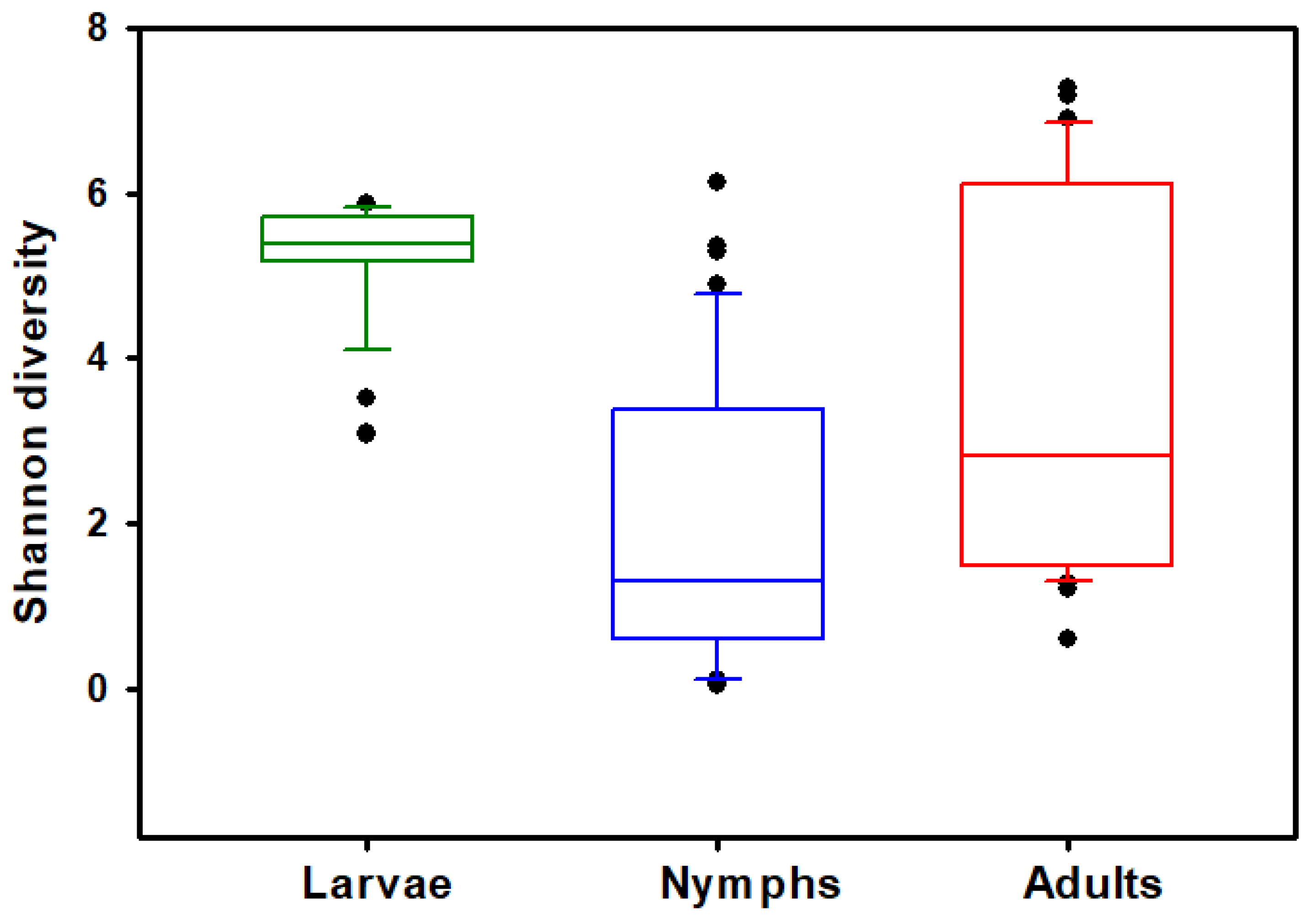
3.2. Alpha Diversity
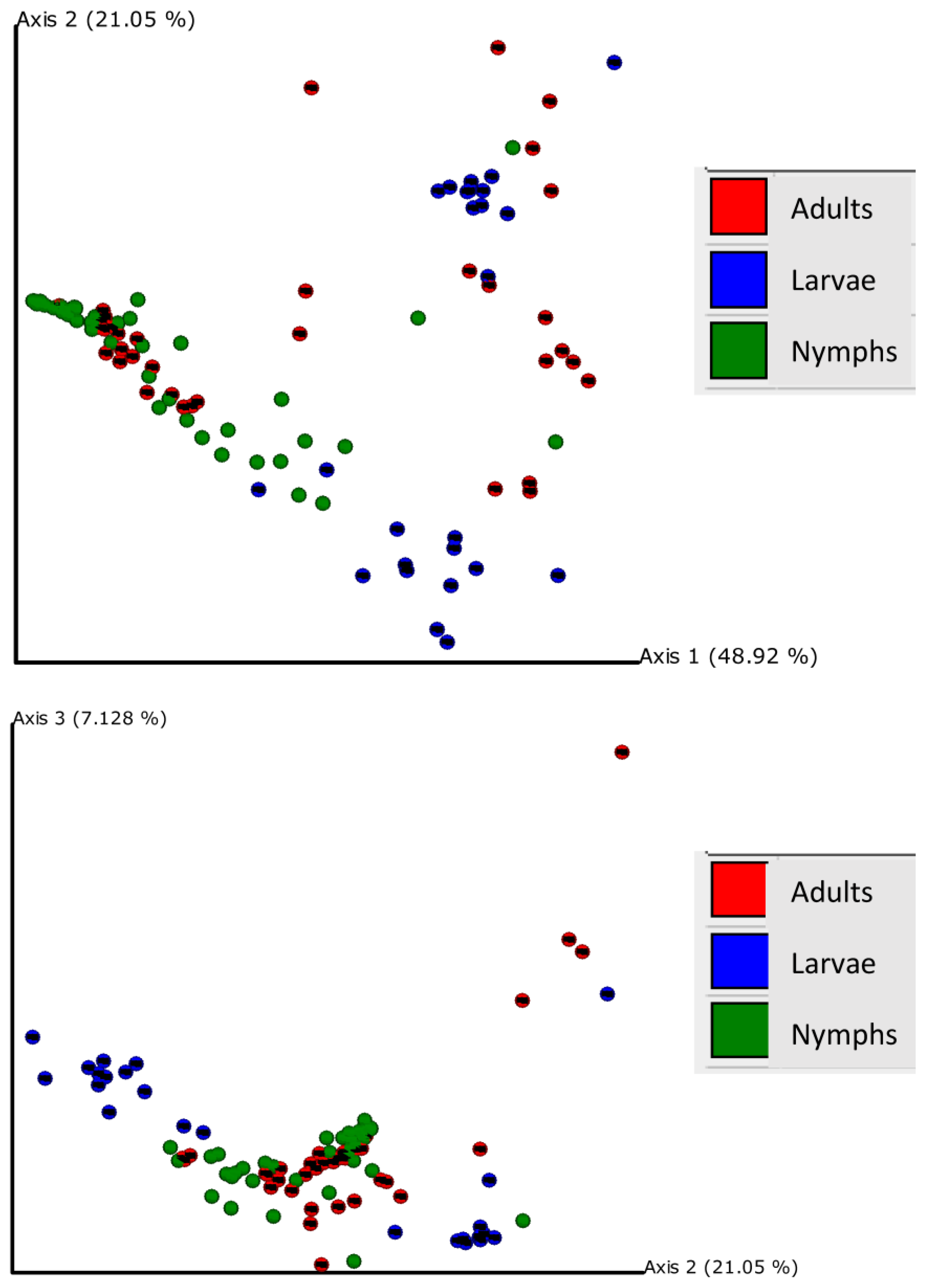
3.3. Beta Diversity
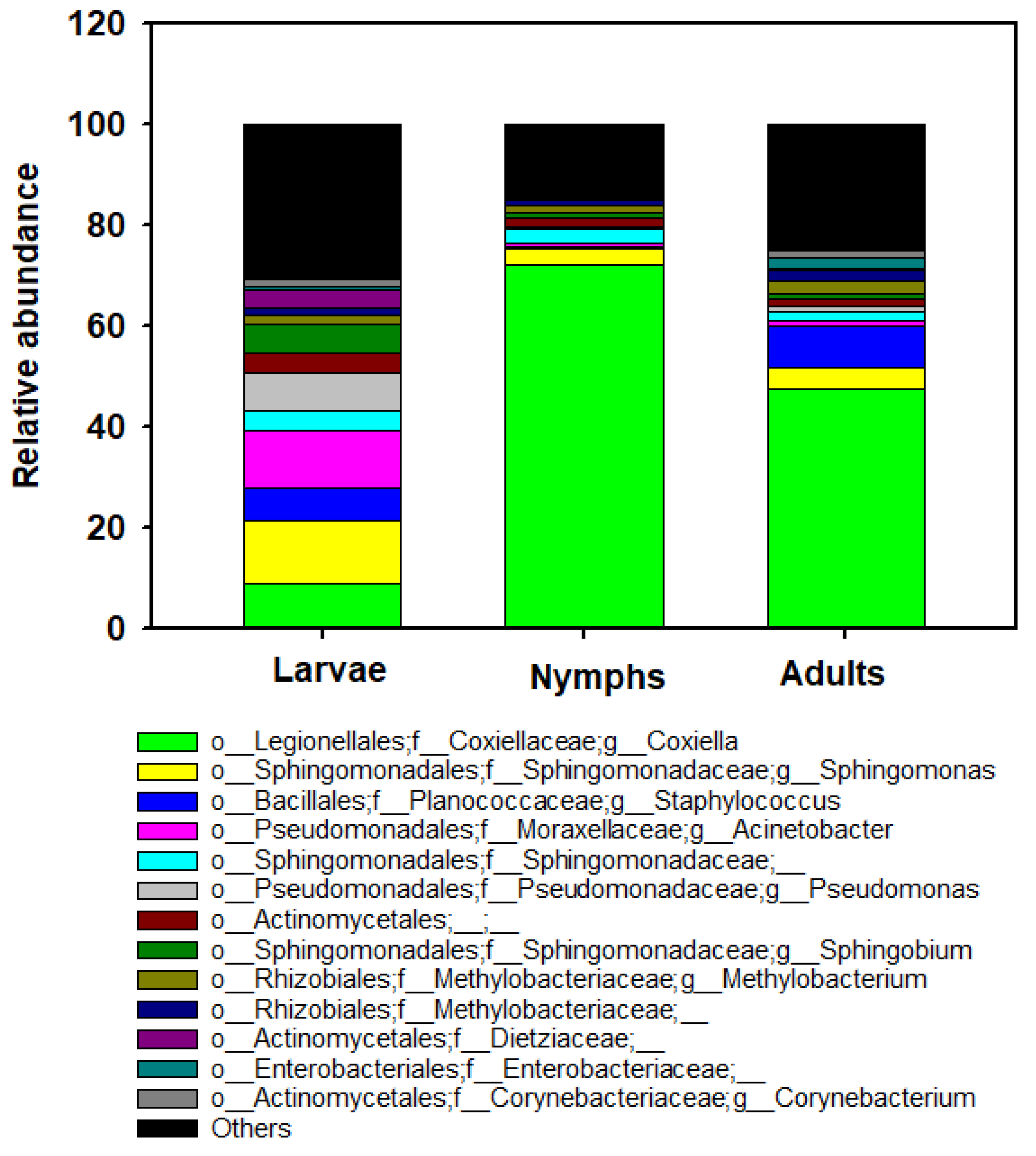
3.4. Bacterial Community Composition
3.5. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- World Health Organization. A Global Brief on Vector-Borne Diseases; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Portillo, A.; Ruiz-Arrondo, I.; Oteo, J.A. Artrópodos vectores en España y sus enfermedades transmisibles. Med. Clin. 2018, 151, 450–459. [Google Scholar] [CrossRef]
- Macaluso, K.; Paddock, C.D.; Sonenshine, D.; Roe, R. Tick-borne spotted fever group rickettsioses and Rickettsia species. Biol. Ticks 2014, 2, 211–250. [Google Scholar]
- Werner, S.L.; Banda, B.K.; Burnsides, C.L.; Stuber, A.J. Zoonosis: Update on existing and emerging vector-borne illnesses in the USA. Curr. Emerg. Hosp. Med. Rep. 2019, 7, 91–106. [Google Scholar] [CrossRef]
- Heath, A. Biology, ecology and distribution of the tick, Haemaphysalis longicornis Neumann (Acari: Ixodidae) in New Zealand. N. Z. Vet. J. 2016, 64, 10–20. [Google Scholar] [CrossRef]
- Luo, L.-M.; Zhao, L.; Wen, H.-L.; Zhang, Z.-T.; Liu, J.-W.; Fang, L.-Z.; Xue, Z.-F.; Ma, D.-Q.; Zhang, X.-S.; Ding, S.-J. Haemaphysalis longicornis ticks as reservoir and vector of severe fever with thrombocytopenia syndrome virus in China. Emerg. Infect. Dis. 2015, 21, 1770. [Google Scholar] [CrossRef]
- Rainey, T.; Occi, J.L.; Robbins, R.G.; Egizi, A. Discovery of Haemaphysalis longicornis (Ixodida: Ixodidae) parasitizing a sheep in New Jersey, United States. J. Med. Entomol. 2018, 55, 757–759. [Google Scholar] [CrossRef]
- Qin, X.-R.; Han, F.-J.; Luo, L.-M.; Zhao, F.-M.; Han, H.-J.; Zhang, Z.-T.; Liu, J.-W.; Xue, Z.-F.; Liu, M.-M.; Ma, D.-Q. Anaplasma species detected in Haemaphysalis longicornis tick from China. Ticks Tick Borne Dis. 2018, 9, 840–843. [Google Scholar] [CrossRef]
- Noguchi, M.; Oshita, S.; Yamazoe, N.; Miyazaki, M.; Takemura, Y.C. Important clinical features of Japanese spotted fever. Am. J. Trop. Med. Hyg. 2018, 99, 466. [Google Scholar] [CrossRef]
- Calisher, C.H. Medically important arboviruses of the United States and Canada. Clin. Microbiol. Rev. 1994, 7, 89–116. [Google Scholar] [CrossRef]
- Hermance, M.E.; Thangamani, S. Powassan virus: An emerging arbovirus of public health concern in North America. Vector Borne Zoonotic Dis. 2017, 17, 453–462. [Google Scholar] [CrossRef]
- Price, K.J.; Graham, C.B.; Witmier, B.J.; Chapman, H.A.; Coder, B.L.; Boyer, C.N.; Foster, E.; Maes, S.E.; Bai, Y.; Eisen, R.J. Borrelia burgdorferi sensu stricto DNA in field-collected Haemaphysalis longicornis ticks, Pennsylvania, United States. Emerg. Infect. Dis. 2021, 27, 608. [Google Scholar] [CrossRef]
- Cumbie, A.N.; Trimble, R.N.; Eastwood, G. Pathogen spillover to an invasive tick species: First detection of Bourbon virus in Haemaphysalis longicornis in the United States. Pathogens 2022, 11, 454. [Google Scholar] [CrossRef]
- Thompson, A.T.; White, S.; Shaw, D.; Egizi, A.; Lahmers, K.; Ruder, M.G.; Yabsley, M.J. Theileria orientalis Ikeda in host-seeking Haemaphysalis longicornis in Virginia, USA. Ticks Tick Borne Dis. 2020, 11, 101450. [Google Scholar] [CrossRef]
- Dye-Braumuller, K.C.; Gual-Gonzalez, L.; Abiodun, T.; Rustin, L.P.; Evans, C.L.; Meyer, M.M.; Zellars, K.; Neault, M.J.; Nolan, M.S. Invasive Haemaphysalis longicornis (Acari: Ixodidae) investigation in South Carolina: New records of establishment, pathogen prevalence, and blood meal analyses. J. Med. Entomol. 2023, 60, 1398–1405. [Google Scholar] [CrossRef]
- Breuner, N.E.; Ford, S.L.; Hojgaard, A.; Osikowicz, L.M.; Parise, C.M.; Rizzo, M.F.R.; Bai, Y.; Levin, M.L.; Eisen, R.J.; Eisen, L. Failure of the Asian longhorned tick, Haemaphysalis longicornis, to serve as an experimental vector of the Lyme disease spirochete, Borrelia burgdorferi sensu stricto. Ticks Tick Borne Dis. 2020, 11, 101311. [Google Scholar] [CrossRef]
- Levin, M.L.; Stanley, H.M.; Hartzer, K.; Snellgrove, A.N. Incompetence of the Asian longhorned tick (Acari: Ixodidae) in transmitting the agent of human granulocytic anaplasmosis in the United States. J. Med. Entomol. 2021, 58, 1419–1423. [Google Scholar] [CrossRef]
- Stanley, H.M.; Ford, S.L.; Snellgrove, A.N.; Hartzer, K.; Smith, E.B.; Krapiunaya, I.; Levin, M.L. The ability of the invasive Asian longhorned tick Haemaphysalis longicornis (Acari: Ixodidae) to acquire and transmit Rickettsia rickettsii (Rickettsiales: Rickettsiaceae), the agent of Rocky Mountain Spotted Fever, under laboratory conditions. J. Med. Entomol. 2020, 57, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.B.; Occi, J.; Bonilla, D.L.; Egizi, A.M.; Fonseca, D.M.; Mertins, J.W.; Backenson, B.P.; Bajwa, W.I.; Barbarin, A.M.; Bertone, M.A. Multistate infestation with the exotic disease–vector tick Haemaphysalis longicornis—United States, August 2017–September 2018. MMWR Morb. Mortal. Wkly. 2018, 67, 1310. [Google Scholar] [CrossRef] [PubMed]
- US Department of Agriculture (USDA). National Haemaphysalis longicornis (Asian Longhorned Tick) Situation Report; USDA: Washington, DC, USA, 2023.
- Rochlin, I.; Benach, J.L.; Furie, M.B.; Thanassi, D.G.; Kim, H.K. Rapid invasion and expansion of the Asian longhorned tick (Haemaphysalis longicornis) into a new area on Long Island, New York, USA. Ticks Tick Borne Dis. 2023, 14, 102088. [Google Scholar] [CrossRef] [PubMed]
- Occi, J.L.; Egizi, A.M.; Robbins, R.G.; Fonseca, D.M. Annotated list of the hard ticks (Acari: Ixodida: Ixodidae) of New Jersey. J. Med. Entomol. 2019, 56, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wormser, G.P.; McKenna, D.; Piedmonte, N.; Vinci, V.; Egizi, A.M.; Backenson, B.; Falco, R.C. First recognized human bite in the United States by the Asian longhorned tick, Haemaphysalis longicornis. Clin. Infe. Dis. 2020, 70, 314–316. [Google Scholar] [CrossRef]
- Bouquet, J.; Melgar, M.; Swei, A.; Delwart, E.; Lane, R.S.; Chiu, C.Y. Metagenomic-based surveillance of Pacific Coast tick Dermacentor occidentalis identifies two novel bunyaviruses and an emerging human ricksettsial pathogen. Sci. Rep. 2017, 7, 12234. [Google Scholar] [CrossRef]
- Tokarz, R.; Lipkin, W.I. Discovery and surveillance of tick-borne pathogens. J. Med. Entomol. 2021, 58, 1525–1535. [Google Scholar] [CrossRef]
- Cross, S.T.; Kapuscinski, M.L.; Perino, J.; Maertens, B.L.; Weger-Lucarelli, J.; Ebel, G.D.; Stenglein, M.D. Co-infection patterns in individual Ixodes scapularis ticks reveal associations between viral, eukaryotic and bacterial microorganisms. Viruses 2018, 10, 388. [Google Scholar] [CrossRef]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol 2015, 31, 315–323. [Google Scholar] [CrossRef]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef]
- Kim, M.; Kim, J.Y.; Yi, M.-H.; Lee, I.-Y.; Yong, D.; Jeon, B.-Y.; Yong, T.-S. Microbiome of Haemaphysalis longicornis tick in Korea. Korean J. Parasitol. 2021, 59, 489–496. [Google Scholar] [CrossRef]
- Egizi, A.M.; Robbins, R.G.; Beati, L.; Nava, S.; Occi, J.L.; Fonseca, D.M. A pictorial key to differentiate the recently detected exotic Haemaphysalis longicornis Neumann, 1901 (Acari, Ixodidae) from native congeners in North America. Zookeys 2019, 818, 117–128. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Gonzalez, A.; Treuren, W.V.; Weiss, S.; Parobek, C.M.; Juliano, J.J.; Knight, R.; Roe, R.M.; Apperson, C.S.; Meshnick, S.R. Diversity of Rickettsiales in the microbiome of the Lone Star tick, Amblyomma americanum. Appl. Environ. Microbiol. 2014, 80, 354–359. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. App. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational multivariate analysis of variance (PERMANOVA). Wiley Statsref Stat. Ref. Online 2014, 1–15. [Google Scholar]
- Anderson, M.; Braak, C.T. Permutation tests for multi-factorial analysis of variance. J. Stat. Comput. Simul. 2003, 73, 85–113. [Google Scholar] [CrossRef]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 2047-217X-2-16. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y. QIIME 2 enables comprehensive end-to-end analysis of diverse microbiome data and comparative studies with publicly available data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 2–3. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Drexler, N.A.; Dahlgren, F.S.; Heitman, K.N.; Massung, R.F.; Paddock, C.D.; Behravesh, C.B. National surveillance of Spotted Fever Group Rickettsioses in the United States, 2008–2012. Am. J. Trop. Med. Hyg. 2016, 94, 26–34. [Google Scholar] [CrossRef]
- Bishop, A.; Borski, J.; Wang, H.-H.; Donaldson, T.G.; Michalk, A.; Montgomery, A.; Heldman, S.; Mogg, M.; Derouen, Z.; Grant, W.E. Increasing incidence of spotted fever group rickettsioses in the United States, 2010–2018. Vector Borne Zoonotic Dis. 2022, 22, 491–497. [Google Scholar] [CrossRef]
- Heitman, K.N.; Dahlgren, F.S.; Drexler, N.A.; Massung, R.F.; Behravesh, C.B. Increasing incidence of ehrlichiosis in the United States: A summary of national surveillance of Ehrlichia chaffeensis and Ehrlichia ewingii infections in the United States, 2008–2012. Am. J. Trop. Med. Hyg. 2016, 94, 52. [Google Scholar] [CrossRef]
- Kwan, J.Y.; Griggs, R.; Chicana, B.; Miller, C.; Swei, A. Vertical vs. horizontal transmission of the microbiome in a key disease vector, Ixodes pacificus. Mol. Ecol. 2017, 26, 6578–6589. [Google Scholar] [CrossRef] [PubMed]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef]
- Zolnik, C.P.; Prill, R.J.; Falco, R.C.; Daniels, T.J.; Kolokotronis, S.O. Microbiome changes through ontogeny of a tick pathogen vector. Mol. Ecol. 2016, 25, 4963–4977. [Google Scholar] [CrossRef] [PubMed]
- Van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; Andreadis, T.G.; Falco, R.C.; Ziegler, L.B.; Hathaway, N. Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef] [PubMed]
- Carpi, G.; Cagnacci, F.; Wittekindt, N.E.; Zhao, F.; Qi, J.; Tomsho, L.P.; Drautz, D.I.; Rizzoli, A.; Schuster, S.C. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS ONE 2011, 6, e25604. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Livanova, N.N.; Fomenko, N.V.; Tupikin, A.E.; Rar, V.A.; Kabilov, M.R.; Livanov, S.G.; Tikunova, N.V. Comparative metagenomic profiling of symbiotic bacterial communities associated with Ixodes persulcatus, Ixodes pavlovskyi and Dermacentor reticulatus ticks. PLoS ONE 2015, 10, e0131413. [Google Scholar] [CrossRef]
- Clow, K.M.; Weese, J.S.; Rousseau, J.; Jardine, C.M. Microbiota of field-collected Ixodes scapularis and Dermacentor variabilis from eastern and southern Ontario, Canada. Ticks Tick Borne Dis. 2018, 9, 235–244. [Google Scholar] [CrossRef]
- Landesman, W.J.; Mulder, K.; Allan, B.F.; Bashor, L.A.; Keesing, F.; LoGiudice, K.; Ostfeld, R.S. Potential effects of blood meal host on bacterial community composition in Ixodes scapularis nymphs. Ticks Tick Borne Dis. 2019, 10, 523–527. [Google Scholar] [CrossRef]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; DePonte, K.; Fish, D.; Fikrig, E. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef]
- Duron, O.; Binetruy, F.; Noël, V.; Cremaschi, J.; McCoy, K.D.; Arnathau, C.; Plantard, O.; Goolsby, J.; Pérez de León, A.A.; Heylen, D.J. Evolutionary changes in symbiont community structure in ticks. Mol. Ecol. 2017, 26, 2905–2921. [Google Scholar] [CrossRef] [PubMed]
- Yessinou, R.E.; Katja, M.-S.; Heinrich, N.; Farougou, S. Prevalence of Coxiella-infections in ticks—Review and meta-analysis. Ticks Tick-Borne Dis. 2022, 13, 101926. [Google Scholar] [CrossRef]
- Körner, S.; Makert, G.R.; Ulbert, S.; Pfeffer, M.; Mertens-Scholz, K. The prevalence of Coxiella burnetii in hard ticks in Europe and their role in Q fever transmission revisited-a systematic review. Front. Vet. Sci. 2021, 8, 655715. [Google Scholar] [CrossRef]
- Nooroong, P.; Trinachartvanit, W.; Baimai, V.; Ahantarig, A. Phylogenetic studies of bacteria (Rickettsia, Coxiella, and Anaplasma) in Amblyomma and Dermacentor ticks in Thailand and their co-infection. Ticks Tick Borne Dis. 2018, 9, 963–971. [Google Scholar] [CrossRef]
- Almeida, A.P.; Marcili, A.; Leite, R.C.; Nieri-Bastos, F.A.; Domingues, L.N.; Martins, J.R.; Labruna, M.B. Coxiella symbiont in the tick Ornithodoros rostratus (Acari: Argasidae). Ticks Tick Borne Dis. 2012, 3, 203–206. [Google Scholar] [CrossRef]
- Jasinskas, A.; Zhong, J.; Barbour, A.G. Highly prevalent Coxiella sp. bacterium in the tick vector Amblyomma americanum. Appl. Environ. Microbiol. 2007, 73, 334–336. [Google Scholar] [CrossRef]
- Duron, O.; Jourdain, E.; McCoy, K.D. Diversity and global distribution of the Coxiella intracellular bacterium in seabird ticks. Ticks Tick Borne Dis. 2014, 5, 557–563. [Google Scholar] [CrossRef]
- Zhang, C.-M.; Li, N.-X.; Zhang, T.-T.; Qiu, Z.-X.; Li, Y.; Li, L.-W.; Liu, J.-Z. Endosymbiont CLS-HI plays a role in reproduction and development of Haemaphysalis longicornis. Exp. App. Acarol. 2017, 73, 429–438. [Google Scholar] [CrossRef]
- Guimard, T.; Amrane, S.; Prudent, E.; El Karkouri, K.; Raoult, D.; Angelakis, E. Case report: Scalp eschar and neck lymphadenopathy associated with bacteremia due to Coxiella-like bacteria. Am. J. Trop. Med. Hyg. 2017, 97, 1319. [Google Scholar] [CrossRef]
- Egyed, L.; Makrai, L. Cultivable internal bacterial flora of ticks isolated in Hungary. Exp. Appl. Acarol. 2014, 63, 107–122. [Google Scholar] [CrossRef]
- Segura, J.A.; Isaza, J.P.; Botero, L.E.; Alzate, J.F.; Gutiérrez, L.A. Assessment of bacterial diversity of Rhipicephalus microplus ticks from two livestock agroecosystems in Antioquia, Colombia. PLoS ONE 2020, 15, e0234005. [Google Scholar] [CrossRef]
- Wang, S.; Hua, X.; Cui, L. Characterization of microbiota diversity of engorged ticks collected from dogs in China. J. Vet. Sci. 2021, 22, e37. [Google Scholar] [CrossRef]
- Campos, B.; Pickering, A.C.; Rocha, L.S.; Aguilar, A.P.; Fabres-Klein, M.H.; de Oliveira Mendes, T.A.; Fitzgerald, J.R.; de Oliveira Barros Ribon, A. Diversity and pathogenesis of Staphylococcus aureus from bovine mastitis: Current understanding and future perspectives. BMC Vet. Res. 2022, 18, 115. [Google Scholar] [CrossRef]
- Von Eiff, C.; Becker, K.; Machka, K.; Stammer, H.; Peters, G. Nasal carriage as a source of Staphylococcus aureus bacteremia. N. Engl. J. Med. 2001, 344, 11–16. [Google Scholar] [CrossRef]
- Cvetnic, L.; Samardzija, M.; Duvnjak, S.; Habrun, B.; Cvetnic, M.; Jaki Tkalec, V.; Duricic, D.; Benic, M. Multi locus sequence typing and spa typing of Staphylococcus aureus Isolated from the milk of cows with subclinical mastitis in croatia. Microorganisms 2021, 9, 725. [Google Scholar] [CrossRef]
- Miranda-Miranda, E.; Cossio-Bayugar, R.; Quezada-Delgado, M.D.R.; Sachman-Ruiz, B.; Reynaud, E. Staphylococcus saprophyticus is a pathogen of the cattle tick Rhipicephalus (Boophilus) microplus. Biocontrol Sci. Technol. 2010, 20, 1055–1067. [Google Scholar] [CrossRef]
- Travanty, N.V.; Ponnusamy, L.; Kakumanu, M.L.; Nicholson, W.L.; Apperson, C.S. Diversity and structure of the bacterial microbiome of the American dog tick, Dermacentor variabilis, is dominated by the endosymbiont Francisella. Symbiosis 2019, 79, 239–250. [Google Scholar] [CrossRef]
- Richardson, E.A.; Roe, R.M.; Apperson, C.S.; Ponnusamy, L. Rickettsia amblyommatis in Ticks: A review of distribution, pathogenicity, and diversity. Microorganisms 2023, 11, 493. [Google Scholar] [CrossRef]
- Lee, S.; Kakumanu, M.L.; Ponnusamy, L.; Vaughn, M.; Funkhouser, S.; Thornton, H.; Meshnick, S.R.; Apperson, C.S. Prevalence of Rickettsiales in ticks removed from the skin of outdoor workers in North Carolina. Parasite Vectors 2014, 7, 607. [Google Scholar] [CrossRef]
- Mixson, T.R.; Campbell, S.R.; Gill, J.S.; Ginsberg, H.S.; Reichard, M.V.; Schulze, T.L.; Dasch, G.A. Prevalence of Ehrlichia, Borrelia, and Rickettsial agents in Amblyomma americanum (Acari: Ixodidae) collected from nine states. J. Med. Entomol. 2006, 43, 1261–1268. [Google Scholar] [CrossRef]
- Jiang, J.; Yarina, T.; Miller, M.K.; Stromdahl, E.Y.; Richards, A.L. Molecular detection of Rickettsia amblyommii in Amblyomma americanum parasitizing humans. Vector Borne Zoonotic Dis. 2010, 10, 329–340. [Google Scholar] [CrossRef]
- Delisle, J.; Mendell, N.L.; Stull-Lane, A.; Bloch, K.C.; Bouyer, D.H.; Moncayo, A.C. Human infections by multiple spotted fever group rickettsiae in Tennessee. Am. J. Trop. Med. Hyg. 2016, 94, 1212. [Google Scholar] [CrossRef]
- Yen, W.-Y.; Stern, K.; Mishra, S.; Helminiak, L.; Sanchez-Vicente, S.; Kim, H.K. Virulence potential of Rickettsia amblyommatis for spotted fever pathogenesis in mice. Pathog. Dis. 2021, 79, ftab024. [Google Scholar] [CrossRef]
- Levin, M.L.; Schumacher, L.B.; Snellgrove, A. Effects of Rickettsia amblyommatis infection on the vector competence of Amblyomma americanum ticks for Rickettsia rickettsii. Vector Borne Zoonotic Dis. 2018, 18, 579–587. [Google Scholar] [CrossRef]
- Rusiñol, M.; Hundesa, A.; Cárdenas-Youngs, Y.; Fernández-Bravo, A.; Pérez-Cataluña, A.; Moreno-Mesonero, L.; Moreno, Y.; Calvo, M.; Alonso, J.L.; Figueras, M.J. Microbiological contamination of conventional and reclaimed irrigation water: Evaluation and management measures. Sci. Total Environ. 2020, 710, 136298. [Google Scholar] [CrossRef]
- Latif-Eugenín, F.; Beaz-Hidalgo, R.; Silvera-Simón, C.; Fernandez-Cassi, X.; Figueras, M.J. Chlorinated and ultraviolet radiation-treated reclaimed irrigation water is the source of Aeromonas found in vegetables used for human consumption. Environmental. Res. 2017, 154, 190–195. [Google Scholar] [CrossRef]
- Pablos, M.; Remacha, M.-A.; Rodríguez-Calleja, J.-M.; Santos, J.; Otero, A.; García-López, M.-L. Identity, virulence genes, and clonal relatedness of Aeromonas isolates from patients with diarrhea and drinking water. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1163–1172. [Google Scholar] [CrossRef]
- Kondo, M.; Matsushima, Y.; Nakanishi, T.; Iida, S.; Habe, K.; Yamanaka, K. Epidemiological study of ticks harbouring Aeromonas hydrophila in areas endemic and non-endemic to Japanese-spotted fever. Trop. Med. Int. Health 2023, 28, 151–156. [Google Scholar] [CrossRef]
- Stojek, N.M.; Dutkiewicz, J. Studies on the occurrence of Gram-negative bacteria in ticks: Ixodes ricinus as a potential vector of Pasteurella. Ann. Agric. Environ. Med. 2004, 11, 319–322. [Google Scholar]
- Ghenghesh, K.S.; Ahmed, S.F.; El-Khalek, R.A.; Al-Gendy, A.; Klena, J. Aeromonas-associated infections in developing countries. J. Infen. Devel. Count. 2008, 2, 081–098. [Google Scholar] [CrossRef]
- Hernández-Jarguín, A.; Díaz-Sánchez, S.; Villar, M.; de la Fuente, J. Integrated metatranscriptomics and metaproteomics for the characterization of bacterial microbiota in unfed Ixodes ricinus. Ticks Tick Borne Dis. 2018, 9, 1241–1251. [Google Scholar] [CrossRef]
- Baldrian, P. Microbial activity and the dynamics of ecosystem processes in forest soils. Curr. Opin. Microbiol. 2017, 37, 128–134. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | n | Average (SE) Observed Features | Average (SE) Shannon’s Diversity |
|---|---|---|---|
| Adults | 36 | 133.0 (14.2) a | 3.608 (0.381) b |
| Nymphs | 44 | 79.3 (9.9) b | 2.037 (0.268) c |
| Larvae | 25 | 129.6 (8.3) a | 5.237 (0.137) a |
| Stage | Counties | n | Average (SE) Observed Features | Average (SE) Shannon’s Diversity |
|---|---|---|---|---|
| Adults | Ashe | 15 | 217.7 (16.0) a | 6.113 (0.237) a |
| Catawba | 7 | 60.1 (4.4) b | 1.339 (0.157) c | |
| Surry | 14 | 78.6 (8.6) b | 2.057 (0.184) b | |
| Nymphs | Buncombe | 12 | 52.4 (14.2) bc | 1.242 (0.468) bc |
| Catawba | 10 | 163.6 (13.3) a | 3.792 (0.270) a | |
| Madison | 2 | 172.5 (4.5) ab | 4.647 (0.709) ab | |
| Surry | 20 | 44.0 (7.5) c | 1.376 (0.317) c | |
| Larvae | Catawba | 10 | 168.8 (9.7) a | 5.415 (0.076) a |
| Surry | 15 | 103.5 (6.0) b | 5.118 (0.221) a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponnusamy, L.; Travanty, N.V.; Watson, D.W.; Seagle, S.W.; Boyce, R.M.; Reiskind, M.H. Microbiome of Invasive Tick Species Haemaphysalis longicornis in North Carolina, USA. Insects 2024, 15, 153. https://doi.org/10.3390/insects15030153
Ponnusamy L, Travanty NV, Watson DW, Seagle SW, Boyce RM, Reiskind MH. Microbiome of Invasive Tick Species Haemaphysalis longicornis in North Carolina, USA. Insects. 2024; 15(3):153. https://doi.org/10.3390/insects15030153
Chicago/Turabian StylePonnusamy, Loganathan, Nicholas V. Travanty, D. Wes Watson, Steven W. Seagle, Ross M. Boyce, and Michael H. Reiskind. 2024. "Microbiome of Invasive Tick Species Haemaphysalis longicornis in North Carolina, USA" Insects 15, no. 3: 153. https://doi.org/10.3390/insects15030153
APA StylePonnusamy, L., Travanty, N. V., Watson, D. W., Seagle, S. W., Boyce, R. M., & Reiskind, M. H. (2024). Microbiome of Invasive Tick Species Haemaphysalis longicornis in North Carolina, USA. Insects, 15(3), 153. https://doi.org/10.3390/insects15030153