
Virus-like Particles from Wolbachia-Infected Cells May Include a Gene Transfer Agent



Abstract
Simple Summary
Abstract
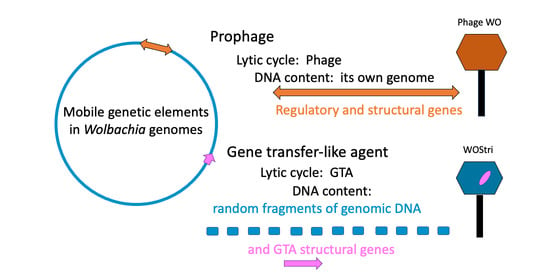
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. DNA Extraction and Purification
2.3. Analysis
3. Results and Discussion
3.1. WOStri Resembles the Proximal Region of Phage WO
3.2. WOStri Gene Order and Sequence Are Conserved in Other WO Phages
3.3. The WOStri Hydrolase Region Contains Evidence for a +1 Programmed Ribosomal Frameshift
3.4. Evidence for Low-Level Packaging of wStri Genomic DNA Encoding EAM-like Proteins
3.5. Evaluation of EAM-like Proteins
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kent, B.N.; Bordenstein, S.R. Phage WO of Wolbachia: Lambda of the endosymbiont world. Trends Microbiol. 2010, 18, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.N.; Funkhouser, L.J.; Setia, S.; Bordenstein, S.R. Evolutionary genetics of a temperate bacteriophage in an obligate intracellular bacteria (Wolbachia). PLoS ONE 2011, 6, e24984. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Bordenstein, S.R. Eukaryotic association module in phage WO genomes from Wolbachia. Nat. Commun. 2016, 7, 13155. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Bordenstein, S.R. Widespread phages of endosymbionts: Phage WO genomics and the proposed taxonomic classification of Symbioviridae. PLoS Genet. 2022, 18, e1010227. [Google Scholar] [CrossRef] [PubMed]
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384. [Google Scholar] [CrossRef]
- Fallon, A.M. DNA recombination and repair in Wolbachia: RecA and related proteins. Mol. Genet. Genom. 2021, 296, 437–456. [Google Scholar] [CrossRef]
- Fallon, A.M.; Baldridge, G.D.; Higgins, L.-A.; Witthuhn, B.A. Wolbachia from the planthopper Laodelphax striatellus establishes a robust, persistent, streptomycin-resistant infection in clonal mosquito cells. In Vitro Cell Dev. Biol. Anim. 2013, 49, 66–73. [Google Scholar] [CrossRef]
- Fallon, A.M. Conditions facilitating infection of mosquito cell lines with Wolbachia, an obligate intracellular bacterium. In Vitro Cell Dev. Biol. Anim. 2019, 55, 120–129. [Google Scholar] [CrossRef]
- Shakya, M.; Soucy, S.M.; Zhaxybayeva, O. Insights into origin and evolution of a-proteobacterial gene transfer agents. Virus Evol. 2017, 3, vex036. [Google Scholar] [CrossRef]
- Lang, A.S.; Zhaxbayeva, O.; Beatty, J.T. Gene transfer agents: Phage-like elements of genetic exchange. Nat. Rev. Microbiol. 2012, 10, 472–482. [Google Scholar] [CrossRef]
- Hynes, A.P.; Mercer, R.G.; Watton, D.E.; Buckley, C.B.; Lang, A.S. DNA packaging bias and differential expression of gene transfer agent genes within a population during production and release of the Rhodobacter capsulatus gene transfer agent, RcGTA. Mol. Microbiol. 2012, 85, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Hynes, A.P.; Shakya, M.; Mercer, R.G.; Grüll, M.P.; Bown, L.; Davidson, F.; Steffen, E.; Matchem, H.; Peach, M.E.; Berger, T.; et al. Functional and evolutionary characterization of a gene transfer agent’s multilocus “genome”. Mol. Biol. Evol. 2016, 33, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Redfield, R.J.; Soucy, S.M. Evolution of bacterial gene transfer agents. Front. Microbiol. 2018, 9, 2527. [Google Scholar] [CrossRef] [PubMed]
- Bárdy, P.; Füzik, T.; Hrebík, D.; Pantůček, R.; Beatty, J.T.; Plevka, P. Structure and mechanism of DNA delivery of a gene transfer agent. Nat. Commun. 2020, 11, 3034. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.; Serbus, L.R. Gene Transfer Agents in Symbiotic Microbes. In Symbiosis: Cellular, Molecular, Medical and Evolutionary Aspects. Results and Problems in Cell Differentiation; Kloc, M., Ed.; Springer: Cham, Switzerland, 2020; Volume 69. [Google Scholar]
- George, E.E.; Tashyreva, D.; Kwong, W.K.; Okamoto, N.; Horák, A.; Husnik, F.; Lukeš, J.; Keeling, P.J. Gene transfer agents in bacterial endosymbionts of microbial eukaryotes. Genome Biol. Evol. 2022, 14, evac099. [Google Scholar] [CrossRef]
- Baldridge, G.D.; Markowski, T.W.; Witthuhn, B.A.; Higgins, L.; Baldridge, A.S.; Fallon, A.M. The Wolbachia WO bacteriophage proteome in the Aedes albopictus C/wStr1 cell line: Evidence for lytic activity? In Vitro Cell Dev. Biol. Anim. 2016, 52, 77–88. [Google Scholar] [CrossRef]
- Masui, S.; Kamoda, S.; Sasaki, T.; Ishikawa, H. Distribution and evolution of bacteriophage WO in Wolbachia, the endosymbiont causing sexual alterations in arthropods. J. Mol. Evol. 2000, 51, 491–497. [Google Scholar] [CrossRef]
- Fallon, A.M. Cytological properties of an Aedes albopictus mosquito cell line infected with Wolbachia strain wAlbB. In Vitro Cell Dev. Biol. Anim. 2008, 44, 154–161. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schaffer, A.A.; Yu, Y.-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Bing, X.L.; Zhao, D.S.; Sun, J.T.; Zhang, J.; Hong, X.Y. Genomic analysis of Wolbachia from Laodelphax striatellus (Delphacidae, Hemiptera) reveals insights into its “Jekyll and Hyde” mode of infection pattern. Genome Biol. Evol. 2020, 12, 3818–3831. [Google Scholar] [CrossRef]
- Fallon, A.M. Muramidase, nuclease, or hypothetical protein genes intervene between paired genes encoding DNA packaging terminase and portal proteins in Wolbachia phages and prophages. Virus Genes 2022, 58, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Mestre, M.R.; Gao, L.A.; Shah, S.A.; López-Beltrán, A.; González-Delgado, A.; Martínez-Abarca, F.; Iranzo, J.; Redrejo-Rodríguez, M.; Zhang, F.; Toro, N. UG/Abi: A highly diverse family of prokaryotic reverse transcriptases associated with defense functions. Nucleic Acid Res. 2022, 50, 6084–6101. [Google Scholar] [CrossRef]
- Labrie, S.J.; Tremblay, D.M.; Moisan, M.; Villion, M.; Magadán, A.H.; Campanacci, V.; Cambillau, C.; Moineau, S. Involvement of the major capsid protein and two early-expressed phage genes in the activity of the lactococcal abortive infection mechanism AbiT. Appl. Environ. Microbiol. 2012, 78, 6890–6899. [Google Scholar] [CrossRef]
- Kampfraath, A.A.; Klasson, L.; Anvar, S.Y.; Vossen, R.H.; Roelofs, D.; Kraaijeveld, K.; Ellers, J. Genome expansion of an obligate parthenogenesis-associated Wolbachia poses an exception to the symbiont reduction model. BMC Genom. 2019, 20, 106. [Google Scholar] [CrossRef]
- Fallon, A.M. Computational evidence for antitoxins associated with RelE/ParE, RatA, Fic, and AbiEii-family toxins in Wolbachia genomes. Mol. Genet. Genom. 2020, 295, 891–909. [Google Scholar] [CrossRef]
- Häuser, R.; Blasche, S.; Dokland, T.; Haggård-Ljungquist, E.; von Brunn, A.; Salas, M.; Casjens, S.; Molineux, I.; Uetz, P. Bacteriophage protein-protein interactions. Adv. Virus Res. 2012, 83, 219–298. [Google Scholar] [CrossRef]
- Christie, G.E.; Calendar, R. Bacteriophage P2. Bacteriophage 2016, 6, e1145782. [Google Scholar] [CrossRef] [PubMed]
- Ghequire, M.G.; De Mot, R. The tailocin tale: Peeling off phage tails. Trends Microbiol. 2015, 23, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Schol, D. Phage tail-like bacteriocins. Annu. Rev. Virol. 2017, 4, 453–467. [Google Scholar] [CrossRef]
- Cianfanelli, F.R.; Alcoforado Diniz, J.; Guo, M.; De Cesare, V.; Trost, M.; Coulthurst, S.J. VgrG and PAAR proteins define distinct versions of a functional Type VI secretion system. PLoS Pathog. 2016, 12, e1005735. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.A.; Funkhouser-Jones, L.J.; Brileya, K.; Reysenbach, A.-L.; Bordenstein, S.R. Antibacterial gene transfer across the tree of life. eLife 2014, 3, e04266. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Sattelberger, E.; Inman, R.B.; Calendar, R.; Loessner, M.J. Genome and proteome of Listeria monocytogenes phage PSA: An unusual case for programmed + 1 translational frameshifting in structural protein synthesis. Mol. Microbiol. 2003, 50, 303–317. [Google Scholar] [CrossRef]
- Sharma, V.; Firth, A.E.; Antonov, I.; Fayet, O.; Atkins, J.F.; Borodovsky, M.; Baranov, P.V. A pilot study of bacterial genes with disrupted orfs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol. Biol. Evol. 2011, 28, 3195–3211. [Google Scholar] [CrossRef] [PubMed]
- Fogg, P.C.M.; Westbye, A.B.; Beatty, J.T. One for all or all for one: Heterogeneous expression and host cell lysis are key to gene transfer agent activity in Rhodobacter capsulatus. PLoS ONE 2012, 7, e43772. [Google Scholar] [CrossRef]
- Matson, E.G.; Thompson, M.G.; Humphrey, S.B.; Zuerner, R.L.; Stanton, T.B. Identification of genes of VSH-1, a prophage-like gene transfer agent of Brachyspira hyodysenteriae. J. Bacteriol. 2005, 187, 5885–5892. [Google Scholar] [CrossRef]
- Zhang, S.; Mao, Y. AAA+ ATPases in protein degradation: Structures, functions and mechanisms. Biomolecules 2020, 10, 629. [Google Scholar] [CrossRef]
- Yedidi, R.S.; Wendler, P.; Enenkel, C. AAA-ATPases in protein degradation. Front. Mol. Biosci. 2017, 4, 42. [Google Scholar] [CrossRef]
- Mahony, J.; Alqarni, M.; Stockdale, S.; Spinelli, S.; Feyereisen, M.; Cambillau, C.; Sinderen, D.V. Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci. Rep. 2016, 8, 36667. [Google Scholar] [CrossRef] [PubMed]
- Belcaid, M.; Bergeron, A.; Poisson, G. The evolution of the tape measure protein: Units, duplications and losses. BMC Bioinform. 2011, 12 (Suppl. 9), S10. [Google Scholar] [CrossRef]
- Zinke, M.; Schröder, G.F.; Lange, A. Major tail proteins of bacteriophages of the order Caudovirales. J. Biol. Chem. 2022, 298, 101472. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.C.; Frank, A.C.; Calteau, A.; Vinnere Pettersson, O.; Granberg, F.; Eriksson, A.S.; Näslund, K.; Holmberg, M.; Lindroos, H.; Andersson, S.G. Run-off replication of host-adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet. 2009, 5, e1000546. [Google Scholar] [CrossRef]
- Guy, L.; Nystedt, B.; Toft, C.; Zaremba-Niedzwiedzka, K.; Berglund, E.C.; Granberg, F.; Naslund, C.; Eriksson, A.-S.; Andersson, S.G.E. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet. 2013, 9, e1003393. [Google Scholar] [CrossRef] [PubMed]
- Québatte, M.; Dehio, C. Bartonella gene transfer agent: Evolution, function, and proposed role in host adaptation. Cell. Microbiol. 2019, 21, e13068. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WOStri | Accessions WP_ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (orf) | wStri | Vit | MUIX_ | wMel_A | wRi_A | wHa_A | wAu_A | wPip_B | wNo_B | wFol_E | WOStri Comments |
| orf1, DNA methylase | 143688966.1 | _02.1 | 010962700.1 | 006280179.1 | 015589002.1 | CDR79043.1 | 007302866.1 | 015588411.1 | 110410402.1 | length varies; N-terminal ParB_N_Srx doman; WOVitA1 ortholog gwv_1137 is query | |
| 143688770.1 | _01.1 | 010962471.1 | 015588907.1 | CDR78683.1 | 007302943.1 | 110410376.1 | |||||
| 143688913.1 | _01.1 | CDR78684.1 | 110410091.1 | ||||||||
| 143688988.1 | _02.1 | ||||||||||
| orf2, HP | 143688769.1 | _01.1 | 010962701.1 | 006280180.1 | 010962472.1 | 010962472.1 | 012481828.1 | 015588412.1 | 110410601.1 | ||
| 143688965.1 | _02.1 | 022626311.1 | 007549853.1 | 015589000.1 | CDR78685.1 | 007302942.1 | 015587745.1 | 110410401.1 | |||
| 143688914.1 | _01.1 | 010962472.1 | 110410092.1 | ||||||||
| 077188538.1 | VitA1 | _01.1, _02.1 | 010082154.1 | ||||||||
| orf3, Ank repeat | 143688768.1 | _01.1 | 010962702.1 | 010082188.1 | 015588999.1 | 010082188.1 | 012481772.1 | 015587744.1 | 110410093.1 | ||
| 143688726.1 | _01.1 | 015588413.1 | 110410375.1 | ||||||||
| 077188539.1 | VitA1 | _01.1 | 110410603.1 | ||||||||
| 143688935.1 | _01.1 | ||||||||||
| 162848928.1 | _01.1 | ||||||||||
| orf4, Terminase | 143688923.1 | _01.1 | 010962703.1 | 012673198.1 | 015588998.1 | 015588914.1 | 012481771.1 | 015588414.1 | 110410400.1 | [26] | |
| 143688915.1 | _01.1 | 010962473.1 | 041582652.1 | 015588914.1 | CDR78687.1 | 015587743.1 | 110410094.1 | ||||
| 077188540.1 | _01.1, _02.1 | 007548331.1 | 110410374.1 | ||||||||
| 241654230.1 | _02.1 | ||||||||||
| orf5, gpW | 143688767.1 | _01.1 | 010962704.1 | 010962704.1 | 010082193.1 | 010082193.1 | 007302777.1 | 015588415.1 | 110410373.1 | [26] | |
| 143688727.1 | _01.1 | 010082193.1 | 007549008.1 | CDR78688.1 | 007301848.1 | 015587742.1 | 110410095.1 | ||||
| 143688830.1 | _01.1 | 007548330.1 | 007302937.1 | 110410399.1 | |||||||
| 077188541.1 | VitA | _01.1, _02.1 | 044475425.1 | ||||||||
| orf 6, Hydrolase | 241654108.1 | _01.1 | 006280957.1 | 015588109.1 | [26]; Figure 3 and Figure 4 | ||||||
| 241654159.1 | _01.1 | 238514242.1 | |||||||||
| 241654099.1 | _01.1 | ||||||||||
| orf7, Hydrolase | 241654107.1 | 006280957.1 | 015588109.1 | ||||||||
| 241654099.1 | 238514242.1 | ||||||||||
| orf8, Portal | 143688977.1 | _02.1 | 012673196.1 | 015588997.1 | CDR78691.1 | 007302936.1 | 015588417.1 | 110410397.1 | [26] | ||
| 096617421.1 | _02.1 | 007549005.1 | CDR79035.1 | 007301845.1 | 110410096.1 | ||||||
| 143688728.1 | _01.1 | 039963649.1 | CDR79034.1 | 012481828.1 | 110410371.1 | ||||||
| 143688827.1 | _01.1 | ||||||||||
| 143688928.1 | _01.1 | ||||||||||
| orf 9, S49 peptidase | 143688824.1 | _01.1 | 006280203.1 | 012673244.1 | 015588996.1 | 006280203.1 | 012481770.1 | 015587741.1 | 110410396.1 | ||
| 006280203.1 | 015588915.1 | CDR79033.1 | 012482035.1 | 110410097.1 | |||||||
| 012481827.1 | 110410370.1 | ||||||||||
| orf 10, Head decoration | 143688823.1 | _01.1 | 010962475.1 | 007548326.1 | 015588917.1 | CDR79031.1 | 012482034.1 | 015588497.1 | 110410369.1 | ||
| 010405448.1 | _01.1 | 010082142.1 | 007549003.1 | 015588995.1 | 010082142.1 | 007302934.1 | 015587740.1 | 110410395.1 | |||
| 143688729.1 | _01.1 | 010082142.1 | 007302870.1 | 110410098.1 | |||||||
| orf 11, Major capsid E | 010405450.1 | VitA,B | _01.1 | 010962707.1 | 007548999.1 | 015588918.1 | 006280204.1 | 012481769.1 | 015587739.1 | 110410368.1 | possible role in abortive infection [27,28,29,30] |
| 143688822.1 | _01.1 | 010962476.1 | 006280204.1 | CDR79030.1 | 012482033.1 | 110410394.1 | |||||
| 007548325.1 | 012481826.1 | 110410579.1 | |||||||||
| 012481801.1 | |||||||||||
| orf 12, HP | 143688821.1 | _01.1 | 007550857.1 | 007550857.1 | 010082171.1 | 007550857.1 | 007301839.1 | 015588264.1 | 110410393.1 | ||
| 143688715.1 | _01.1 | 010082171.1 | 012673243.1 | 010082171.1 | 012481768.1 | 015587738.1 | 110410099.1 | ||||
| 143688795.1 | _01.1 | 007548323.1 | CAQ54530.1 | 110410367.1 | |||||||
| 143688961.1 | _02.1 | 007302873.1 | |||||||||
| 012481800.1 | |||||||||||
| orf13, Tail Z | 143688820.1 | _01.1 | 038228285.1 | 006280676.1 | 010962479.1 | 010962479.1 | 012481823.1 | 015588265.1 | 110410392.1 | Head–tail joining [31,32] | |
| 143688765.1 | _01.1 | 010962479.1 | 006280854.1 | 006280676.1 | 007302874.1 | 015587737.1 | 110410100.1 | ||||
| 143688714.1 | _01.1 | 007301838.1 | 110410366.1 | ||||||||
| 143688794.1 | _01.1 | 007302931.1 | |||||||||
| 143688960.1 | _02.1 | ||||||||||
| 010405454.1 | VitA1 | _01.1 | |||||||||
| ore 14, HP | 143688818.1 | _01.1 | 010962480.1 | 010962732.1 | 015588919.1 | 010962732.1 | 007301837.1 | 015588266.1 | 110410101.1 | ||
| 143688713.1 | _01.1 | 010962732.1 | 006280893.1 | CDR79025.1 | 007302875.1 | 041581447.1 | 110410365.1 | ||||
| 010405456.1 | _01.1 | 012481799.1 | 110410391.1 | ||||||||
| 143688793.1 | _01.1, _02.1 | ||||||||||
| 143688964.1 | _02.1 | ||||||||||
| orf15, baseplate V | 015588267.1 | _01.1 | 010962731.1 | 012673195.1 | 015588920.1 | 012673195.1 | 007302876.1 | 015588267.1 | 110410390.1 | tailocins [33,34,35] | |
| 143688792.1 | _01.1, _02.1 | 007548312.1 | CDR79024.1 | 012481798.1 | 015587735.1 | 110410575.1 | |||||
| 096641463.1 | _01.1 | 007548997.1 | 012482032.1 | 110410102.1 | |||||||
| 143688963.1 | _02.1 | 110410364.1 | |||||||||
| orf 16, PAAR | 143688817.1 | _01.1 | 010082105.1 | 010082105.1 | 015588921.1 | 010082105.1 | 012481797.1 | 015588268.1 | 110410103.1 | [10,33,34,35] | |
| 143688962.1 | _02.1 | 010082109.1 | 007548996.1 | 010082109.1 | 007302877.1 | 015587734.1 | 110410389.1 | ||||
| 063630987.1 | _01.1, _02.1 | 049749693.1 | 012482031.1 | 110410000.1 | |||||||
| 010405460.1 | VitA1 | _01.1 | |||||||||
| 143688712.1 | _01.1 | ||||||||||
| orf 17, Baseplate W | 143688711.1 | _01.1 | 007552425.1 | 007548437.1 | 015588922.1 | 007552425.1 | 007302878.1 | 015588269.1 | 110410104.1 | ||
| 010405462.1 | VitA1 | _01.1 | 015588922.1 | 007552425.1 | 015588922.1 | 012481796.1 | 015588498.1 | 110409999.1 | |||
| 012673242.1 | |||||||||||
| orf 18, Baseplate J | 143688710.1 | _01.1 | 010962482.1 | 012673402.1 | 015588994.1 | 010962482.1 | 012481795.1 | 015588270.1 | 110409998.1 | ||
| 143688959.1 | _02.1 | 010962729.1 | 012673194.1 | 015588923.1 | 012673194.1 | 007301818.1 | 015587733.1 | 110410388.1 | |||
| 143688789.1 | _01.1 | 012673194.1 | 012673241.1 | 012481767.1 | 015588499.1 | 110410105.1 | |||||
| orf 19, Tail protein I | 143688764.1 | _01.1 | 015588924.1 | 012673401.1 | 015588993.1 | CDR78703.1 | 007302879.1 | 015588271.1 | 110409997.1 | ||
| 143688709.1 | _01.1 | 010962728.1 | 006280238.1 | 015588924.1 | CDR79020.1 | 015587732.1 | AWW50841.1 | ||||
| 143688958.1 | _02.1 | 012673240.1 | 015588500.1 | ||||||||
| 077188546.1 | VitA1 | _01.1 | |||||||||
| 143688788.1 | _01.1 | ||||||||||
| ore 20, HP | 143688763.1 | _01.1 | 041580639.1 | 006280237.1 | 015588992.1 | CDR78705.1 | 007302881.1 | 015587731.1 | |||
| 143688865.1 | _01.1 | 044475433.1 | 015588272.1 | ||||||||
| 143688708.1 | _01.1 | ||||||||||
| 177429427.1 | _01.1 | ||||||||||
| 177429437.1 | _02.1 | ||||||||||
| orf 21, HP | 019236494.1 | _01.1 | 010962726.1 | 007549613.1 | 015588273.1 | CDR78707.1 | 007302927.1 | 015587730.1 | |||
| 143688707.1 | _01.1 | 007302882.1 | 015588273.1 | ||||||||
| 143688864.1 | _01.1 | ||||||||||
| 143688957.1 | _02.1 | ||||||||||
| orf 22, DUF 2924 | 143688762.1 | _01.1 | 010962725.1 | 044471239.1 | CDR78708.1 | 007302926.1 | 015587729.1 | 110409343.1 | |||
| 143688785.1 | _01.1 | 010962486.1 | 015588926.1 | CDR79016.1 | 007302883.1 | 110409994.1 | |||||
| 143688706.1 | _01.1 | 015588926.1 | 110409273.1 | ||||||||
| 110410108.1 |
| Copies | Copies | Coverage/Identity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Contig Number | Length | Coverage | Orf Length | wStri Protein Accession | Coverage (%) | Identity (%) | Function | MUIX | Non-MUIX | <50%/50% |
| 2016 data | ||||||||||
| node 1 | 15,817 | 27.7 | 2253 | WP_143688936.1 01.1 | 92 | 99 | AAA-ATPase | 2 | ||
| node 2 | 14,479 | 59.8 | 11,826 | WP_063631194.1 LRUH | 100 | 100 | Ankyrin | 1 | ||
| node 3 | 13,399 | 17.5 | 5772 | WP_206663913.1 01.1 | 98 | 100 | Ankyrin | 1 | ||
| node 4 | 11,891 | 19.2 | 2151 | WP_063630525.1 LRUH | 98 | 100 | AAA-ATPase | 1 | ||
| node 5 | 10,094 | 10.9 | 2307 | WP_143688736.1 01.1 | 99 | 99 | tape measure | 8 | ||
| node 6 | 9405 | 9.9 | 3429 | WP_143688778.1 01.1 | 98 | 81 | Ankyrin | 2 | many <50% | |
| node 7 | 9118 | 3.9 | 5730 | WP_143688882.1 01.1 | 100 | 100 | Ankyrin | 2 | ||
| node 8 | 7970 | 37.5 | 2439 | WP_143688947.1 02.1 | 99 | 100 | tape measure | 8 | ||
| 2020 data | ||||||||||
| node 1 | 36,552 | 184 | 2253 | WP_063630523.1 01.1 | 92 | 100 | AAA-ATPase | 2 | ||
| 2316 | WP_143688796.1 01.1 | 99 | 100 | tape measure | 8 | |||||
| 2601 | WP_063630621.1 02.1 | 99 | 100 | ABC transporter ATP-binding protein/permease | 1 | many <50% | ||||
| node 2 | 30,125 | 397 | 2439 | WP_143688947.1 02.1 | 99 | 100 | tape measure | 8 | ||
| 11826 | WP_063631194.1 LRUH | 100 | 100 | Ankyrin | 1 | |||||
| node 3 | 23,532 | 7 | 2940 | WP_063630441.1 01.1 | 97 | 100 | DUF 3971 | 1 | ||
| node 4 | 19,389 | 153 | 9243 | WP_143688758.1 01.1 | 100 | 100 | Ankyrin | 2 | many <50% | |
| node 5 | 18,215 | 40.8 | 2262 | WP_063631186.1 01.1 | 98 | 100 | HP | 1 | ||
| 3696 | WP_177429433.1 01.1 | 98 | 100 | Collagen-like protein | 2 | |||||
| 5730 | WP_143688882.1 01.1 | 100 | 100 | Ankyrin | 2 | |||||
| node 6 | 17,355 | 5.8 | 2712 | WP_172793444.1 02.1 | 99 | 100 | RecB | 1 | ||
| 3090 | WP_063630611.1 02.1 | 99 | 100 | AIR synthase | 1 | |||||
| node 7 | 15,996 | 4.5 | 8520 | WP_084240976.1 01.1 | 100 | 100 | ββ’ RNA Polymerase | 1 | ||
| node 8 | 14,963 | 148 | 4470 | WP_063631092.1 02.1 | 99 | 100 | Ankyrin | 1 | many <50% | |
| node 9 | 14,237 | 10.5 | 2553 | WP_063630489.1 01.1 | 99 | 100 | ATP-dependent chaperone | 2 | ||
| 4671 | WP_063630487.1 01.1 | 99 | 100 | NAD-glutamate dehydrogenase | 1 | |||||
| node 12 | 12,174 | 6.9 | 2322 | WP_063631238.1 01.1 | 99 | 100 | Translational initiation factor 2 | 1 | ||
| node 13 | 11,829 | 155 | 4245 | WP_143688778.1 01.1 | 98 | 80 | Ankyrin | 2 | many <50% | |
| node 15 | 10,901 | 4 | 2121 | WP_143688815.1 01.1 | 90 | 95 | DNAJ/Ankyrin | 1 | ||
| node 16 | 10,612 | 251 | 2151 | WP_063630525.1 LRUH | 98 | 100 | AAA-ATPase | 1 | ||
| node 17 | 10,406 | 5.4 | 2814 | WP_063631210.1 01.1 | 99 | 100 | EXI ABC subunit UvrA | 1 | ||
| node 18 | 9228 | 18.9 | 3087 | WP_143688902.1 01.1 | 99 | 100 | Ankyrin | 2 | many <50% | |
| node 20 | 8085 | 65.2 | 7416 | WP_143688882.1 01.1 | 99 | 100 | Ankyrin | 2 | ||
| node 24 | 7238 | 3.6 | 3462 | WP_241654150.1 01.1 | 91 | 100 | DEAD/DEAH box helicase | 1 | ||
| node 26 | 6544 | 16.4 | 4368 | WP_143688901.1 01.1 | 99 | 100 | Ankyrin | 1 | ||
| node 28 | 6264 | 274 | 5772 | WP_206663913.1 01.1 | 98 | 100 | Ankyrin | 1 | many <50% | |
| node 34 | 5542 | 36 | 2175 | WP_082246120.1 02.1 | 97 | 100 | AAA-ATPase | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fallon, A.M.; Carroll, E.M. Virus-like Particles from Wolbachia-Infected Cells May Include a Gene Transfer Agent. Insects 2023, 14, 516. https://doi.org/10.3390/insects14060516
Fallon AM, Carroll EM. Virus-like Particles from Wolbachia-Infected Cells May Include a Gene Transfer Agent. Insects. 2023; 14(6):516. https://doi.org/10.3390/insects14060516
Chicago/Turabian StyleFallon, Ann M., and Elissa M. Carroll. 2023. "Virus-like Particles from Wolbachia-Infected Cells May Include a Gene Transfer Agent" Insects 14, no. 6: 516. https://doi.org/10.3390/insects14060516
APA StyleFallon, A. M., & Carroll, E. M. (2023). Virus-like Particles from Wolbachia-Infected Cells May Include a Gene Transfer Agent. Insects, 14(6), 516. https://doi.org/10.3390/insects14060516