
Positive Correlation of the Gene Rearrangements and Evolutionary Rates in the Mitochondrial Genomes of Thrips (Insecta: Thysanoptera)



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Mitogenome Acquisition
2.2. Mitogenome Annotation and Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
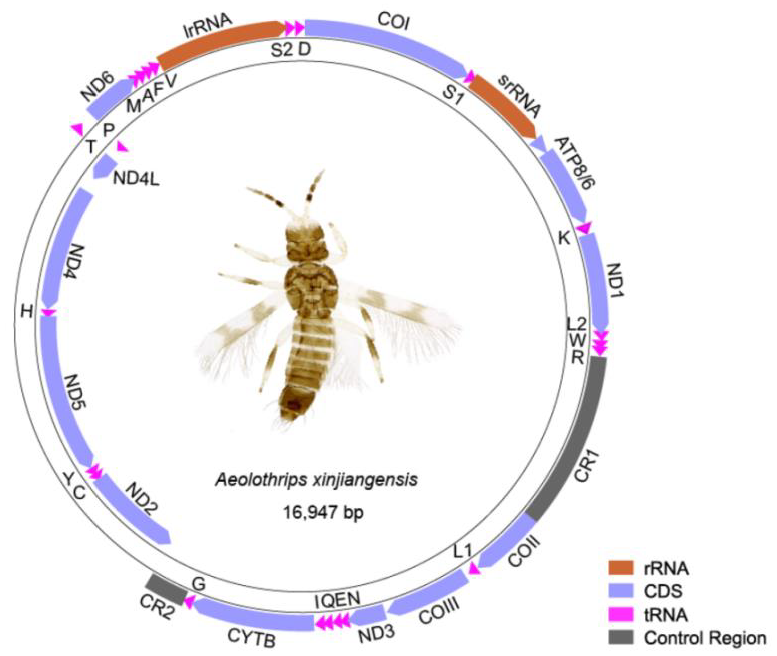
3.1. Mitogenome Feature and Composition
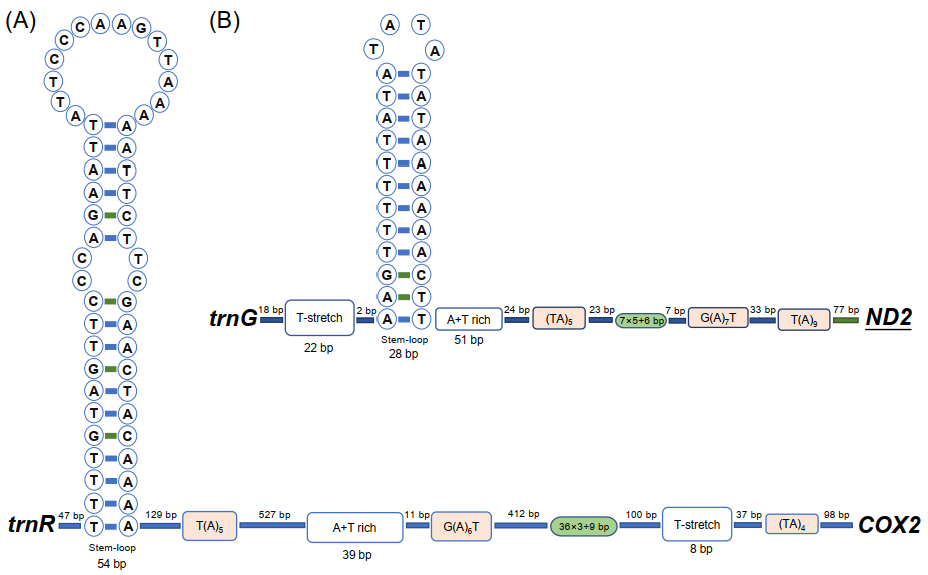
3.2. Non-Coding Regions
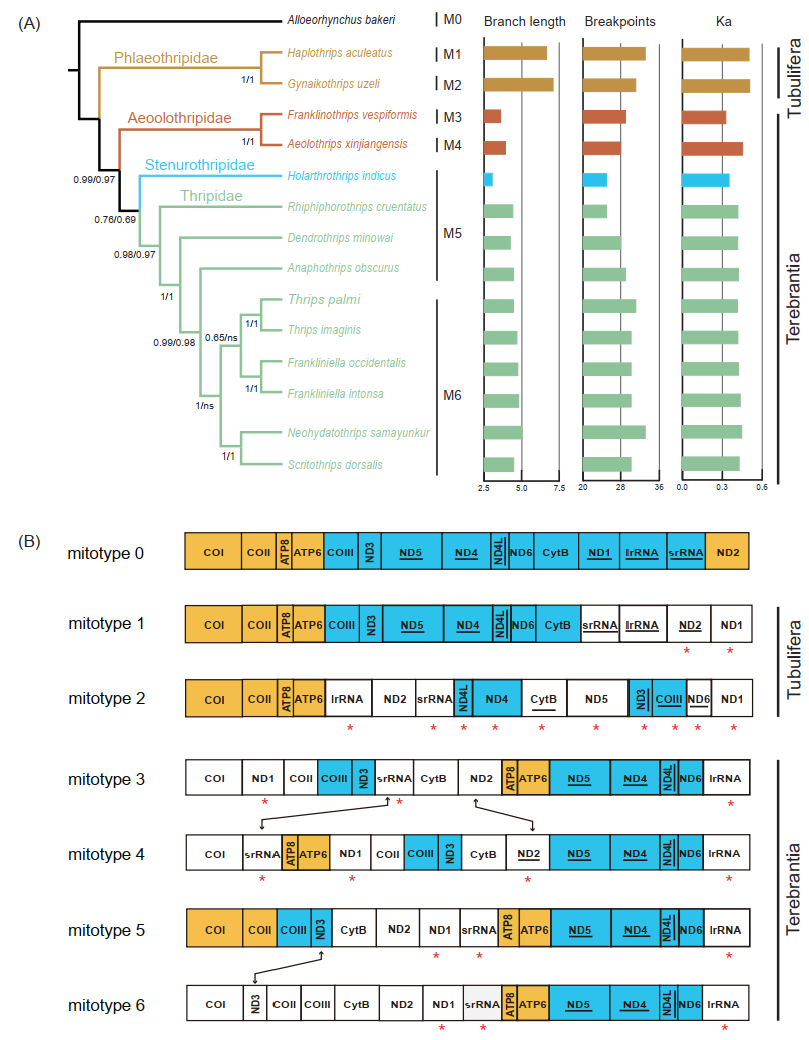
3.3. Gene Rearrangements in the Mitogenome of Banded Thrips
3.4. Correlation between the Degree of Gene Rearrangement and the Evolutionary Rate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2009, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shao, R.; Song, F.; Zhou, X.; Yang, Q.; Li, Z.; Cai, W. Mitochondrial genomes of two barklice, Psococerastis albimaculata and Longivalvus hyalospilus (Psocoptera: Psocomorpha): Contrasting rates in mitochondrial gene rearrangement between major lineages of Psocodea. PLoS ONE 2013, 8, e61685. [Google Scholar] [CrossRef]
- Nie, Y.; Fu, Y.T.; Zhang, Y.; Deng, Y.P.; Wang, W.; Tu, Y.; Liu, G.H. Highly rearranged mitochondrial genome in Falcolipeurus lice (Phthiraptera: Philopteridae) from endangered eagles. Parasites Vectors 2021, 14, 269. [Google Scholar] [CrossRef]
- Shao, R.; Kirkness, E.F.; Barker, S.C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009, 19, 904–912. [Google Scholar] [CrossRef]
- Sweet, A.D.; Johnson, K.P.; Cao, Y.; de Moya, R.S.; Skinner, R.K.; Tan, M.; Virrueta Herrera, S.; Cameron, S.L. Structure, gene order, and nucleotide composition of mitochondrial genomes in parasitic lice from Amblycera. Gene 2021, 768, 145312. [Google Scholar] [CrossRef]
- Chakraborty, R.; Tyagi, K.; Kundu, S.; Rahaman, I.; Singha, D.; Chandra, K.; Patnaik, S.; Kumar, V. The complete mitochondrial genome of melon thrips, Thrips palmi (Thripinae): Comparative analysis. PLoS ONE 2018, 13, e0199404. [Google Scholar] [CrossRef]
- Tyagi, K.; Chakraborty, R.; Cameron, S.L.; Sweet, A.D.; Chandra, K.; Kumar, V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci. Rep. 2020, 10, 695. [Google Scholar] [CrossRef]
- Yan, D.; Tang, Y.; Hu, M.; Liu, F.; Zhang, D.; Fan, J. The mitochondrial genome of Frankliniella intonsa: Insights into the evolution of mitochondrial genomes at lower taxonomic levels in Thysanoptera. Genomics 2014, 104, 306–312. [Google Scholar] [CrossRef]
- Macey, J.R.; Larson, A.; Ananjeva, N.B.; Fang, Z.; Papenfuss, T.J. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 1997, 14, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef] [PubMed]
- San Mauro, D.; Gower, D.J.; Zardoya, R.; Wilkinson, M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 2006, 23, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003, 20, 362–370. [Google Scholar] [CrossRef]
- Yan, D.; Tang, Y.; Xue, X.; Wang, M.; Liu, F.; Fan, J. The complete mitochondrial genome sequence of the western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae) contains triplicate putative control regions. Gene 2012, 506, 117–124. [Google Scholar] [CrossRef]
- Mirab-balou, M.; Tong, X.; Feng, J.; Chen, X. Thrips (Insecta: Thysanoptera) of China. Check List 2011, 7, 720. [Google Scholar] [CrossRef]
- Masumoto, M.; Okajima, S. Review of the Aeolothripidae (Thysanoptera) in Japan. Zootaxa 2019, 4546, 301–326. [Google Scholar] [CrossRef]
- Mound, L.A.; Reynaud, P. Franklinothrips; a pantropical Thysanoptera genus of ant-mimicking obligate predators (Aeolothripidae). Zootaxa 2005, 864, 1–16. [Google Scholar] [CrossRef][Green Version]
- Reynaud, P. Thrips (Thysanoptera). Chapter 13.1. BioRisk 2010, 4, 767–791. [Google Scholar] [CrossRef]
- Han, Y.F.; Pan, Y.C.; Hu, X.G. A new species and three new records of Aeolothrips Haliday from China (Thysanoptera: Aeolothripidae). Acta Zootaxonomica Sin. 1987, 12, 303–306. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Gillett, C.P.D.T.; Crampton-Platt, A.; Timmermans, M.J.T.N.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, T.M. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Blanchette, M.; Kunisawa, T.; Sankoff, D. Gene order breakpoint evidence in animal mitochondrial phylogeny. J. Mol. Evol. 1999, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Dowton, M.; Murrell, A.; Barker, S.C. Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol. Biol. Evol. 2003, 20, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef]
- Julio, R.; Sánchez-DelBarri, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2004, 19, 2496–2497. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Li, H.; Shao, R.; Song, N.; Song, F.; Jiang, P.; Li, Z.; Cai, W. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Clemente, P.; Calvo-Garrido, J.; Maffezzini, C.; Felser, A.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. Mitochondrial polyadenylation is a one-step process required for mRNA integrity and tRNA maturation. PLoS Genet. 2016, 12, e1006028. [Google Scholar] [CrossRef]
- Ji, H.; Xu, X.; Jin, X.; Yin, H.; Luo, J.; Liu, G.; Zhao, Q.; Chen, Z.; Bu, W.; Gao, S. Using high-resolution annotation of insect mitochondrial DNA to decipher tandem repeats in the control region. RNA Biol. 2019, 16, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Toompuu, M.; Tuomela, T.; Laine, P.; Paulin, L.; Dufour, E.; Jacobs, H.T. Polyadenylation and degradation of structurally abnormal mitochondrial tRNAs in human cells. Nucleic Acids Res. 2018, 46, 5209–5226. [Google Scholar] [CrossRef]
- Zhou, Z.; Huang, Y.; Shi, F. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A+T-rich region of 70 bp in length. Genome 2007, 50, 855–866. [Google Scholar] [CrossRef]
- Lewis, D.L.; Farr, C.L.; Kaguni, L.S. Drosophila melanogaster mitochondrial DNA: Completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 1995, 4, 263–278. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Lunt, D.H.; Whipple, L.E.; Hyman, B.C. Mitochondrial DNA variable number tandem repeats (VNTRs): Utility and problems in molecular ecology. Mol. Ecol. 1998, 7, 1441–1455. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Ren, Y.; Sun, Y.; Wu, Z.; Ruan, J.; He, B.; Zhang, T.; Yu, X.; Tian, X.; Bu, W. PacBio full-length transcriptome profiling of insect mitochondrial gene expression. RNA Biol. 2016, 13, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zheng, B.Y.; Zhu, J.C.; van Achterberg, C.; Tang, P.; Chen, X.X. The first two mitochondrial genomes of wood wasps (Hymenoptera: Symphyta): Novel gene rearrangements and higher-level phylogeny of the basal hymenopterans. Int. J. Biol. Macromol. 2019, 123, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Margam, V.M.; Coates, B.S.; Hellmich, R.L.; Agunbiade, T.; Seufferheld, M.J.; Sun, W.; Ba, M.N.; Sanon, A.; Binso-Dabire, C.L.; Baoua, I.; et al. Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS ONE 2011, 6, e16444. [Google Scholar] [CrossRef] [PubMed]
- Buckman, R.S.; Mound, L.A.; Whiting, M.F. Phylogeny of thrips (Insecta: Thysanoptera) based on five molecular loci. Syst. Entomol. 2013, 38, 123–133. [Google Scholar] [CrossRef]
- Zhang, S.M.; Mound, L.; Feng, J.N. Morphological phylogeny of Thripidae (Thysanoptera: Terebrantia). Invertebr. Syst. 2019, 33, 671–696. [Google Scholar] [CrossRef]
- Arribas, P.; Andujar, C.; Moraza, M.L.; Linard, B.; Emerson, B.C.; Vogler, A.P. Mitochondrial metagenomics reveals the ancient origin and phylodiversity of soil mites and provides a phylogeny of the Acari. Mol. Biol. Evol. 2020, 37, 683–694. [Google Scholar] [CrossRef]
- Xue, X.F.; Guo, J.F.; Dong, Y.; Hong, X.Y.; Shao, R. Mitochondrial genome evolution and tRNA truncation in Acariformes mites: New evidence from eriophyoid mites. Sci. Rep. 2016, 6, 18920. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Suborder/Order | Family | Species Name | GenBank Number |
|---|---|---|---|
| Hemiptera | Hemiptera | Alloeorhynchus bakeri | NC_016432 |
| Terebrantia | Aeolothripidae | Aeolothrips xinjiangensis * | MW376485 |
| Franklinothrips vespiformis | MN072395 | ||
| Stenurothripidae | Holarthrothrips indicus | MN072397 | |
| Thripidae | Rhiphiphorothrips cruentatus | MN072396 | |
| Neohydatothrips samayunkur | MF991901 | ||
| Anaphothrips obscurus | NC_035510 | ||
| Thrips palmi | NC_039437 | ||
| Thrips imaginis | AF335993 | ||
| Frankliniella intonsa | JQ917403 | ||
| Frankliniella occidentalis | NC_018370 | ||
| Scritothrips dorsalis | NC_025241 | ||
| Dendrothrips minowai | NC_037839 | ||
| Tubulifera | Phlaeothripidae | Haplothrips aculeatus | NC_027488 |
| Gynaikothrips uzeli | MK940484 |
| Pearson | Spearman | ||
|---|---|---|---|
| Breakpoints | Ka | Branch length | |
| r | 0.563 | 0.831 | |
| p (2-tailed) | 0.036 | 0.000 | |
| N | 14 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; He, J.; Song, F.; Tian, L.; Cai, W.; Li, H. Positive Correlation of the Gene Rearrangements and Evolutionary Rates in the Mitochondrial Genomes of Thrips (Insecta: Thysanoptera). Insects 2022, 13, 585. https://doi.org/10.3390/insects13070585
Liu Q, He J, Song F, Tian L, Cai W, Li H. Positive Correlation of the Gene Rearrangements and Evolutionary Rates in the Mitochondrial Genomes of Thrips (Insecta: Thysanoptera). Insects. 2022; 13(7):585. https://doi.org/10.3390/insects13070585
Chicago/Turabian StyleLiu, Qiaoqiao, Jia He, Fan Song, Li Tian, Wanzhi Cai, and Hu Li. 2022. "Positive Correlation of the Gene Rearrangements and Evolutionary Rates in the Mitochondrial Genomes of Thrips (Insecta: Thysanoptera)" Insects 13, no. 7: 585. https://doi.org/10.3390/insects13070585
APA StyleLiu, Q., He, J., Song, F., Tian, L., Cai, W., & Li, H. (2022). Positive Correlation of the Gene Rearrangements and Evolutionary Rates in the Mitochondrial Genomes of Thrips (Insecta: Thysanoptera). Insects, 13(7), 585. https://doi.org/10.3390/insects13070585