Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Colony
2.2. RNA Extraction and cDNA Library Construction
2.3. Gene Annotation and Mapping
2.4. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Pathway Analyses
3. Results
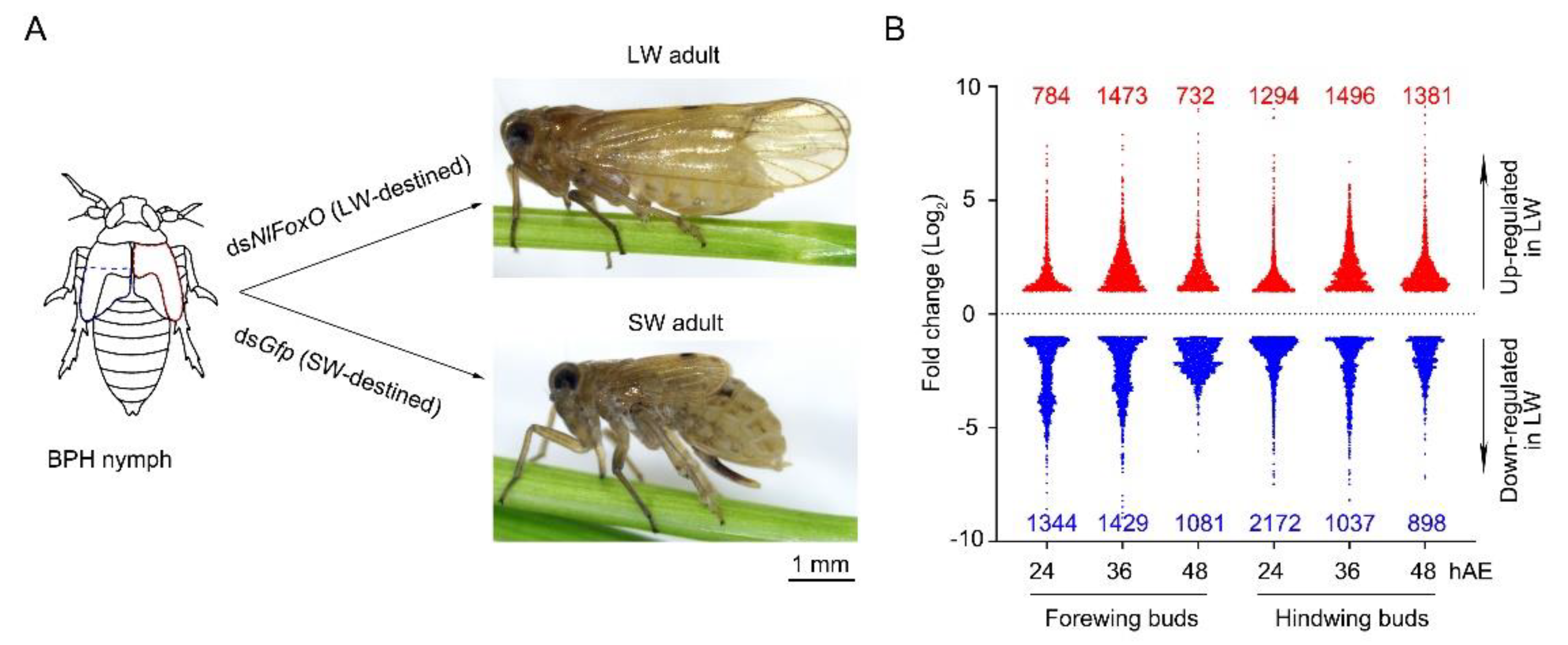
3.1. Overview of Transcriptomes
3.2. DEGs in Wing Buds of 5th-Instar Nymphs Treated with dsNlFoxO and dsGfp
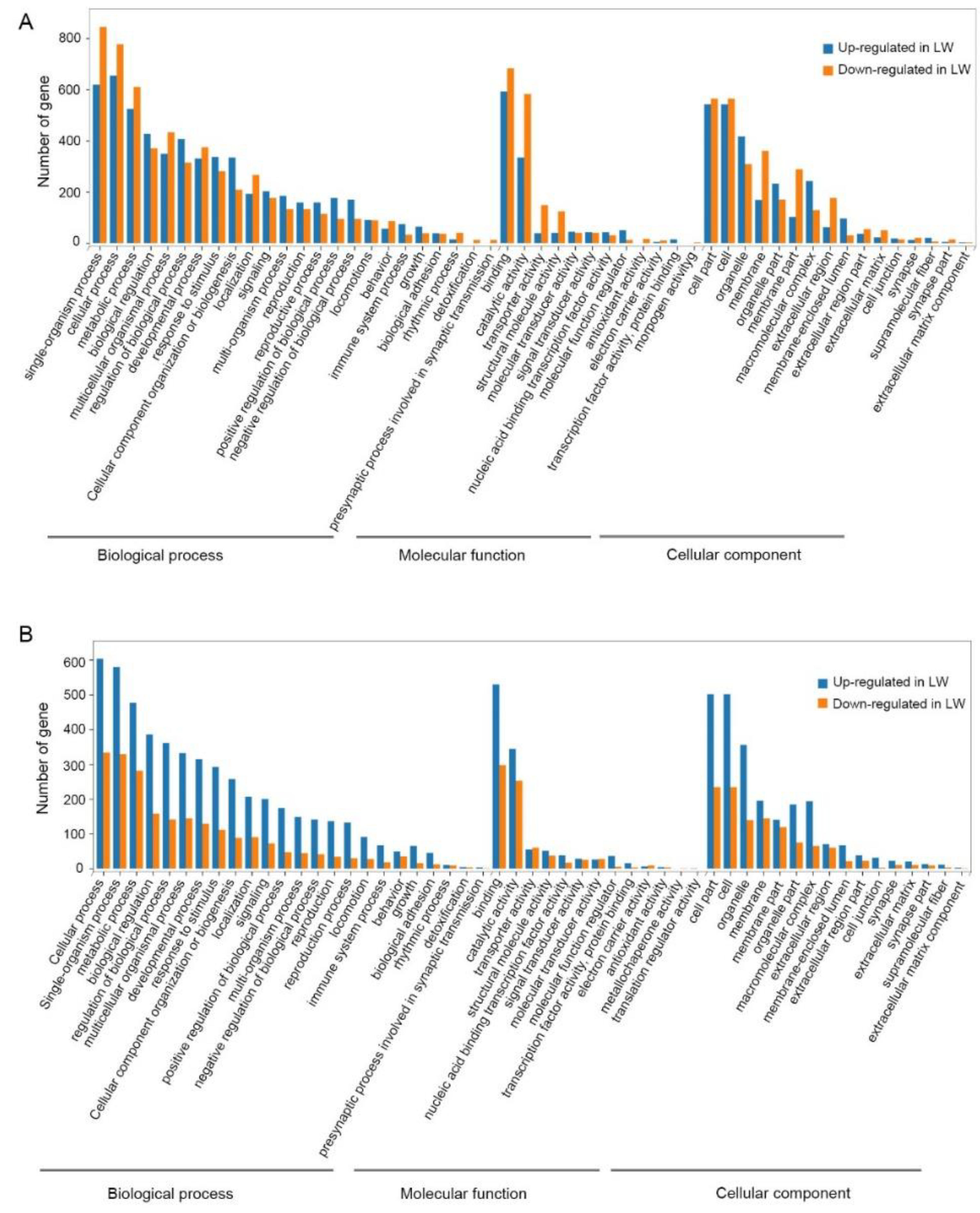
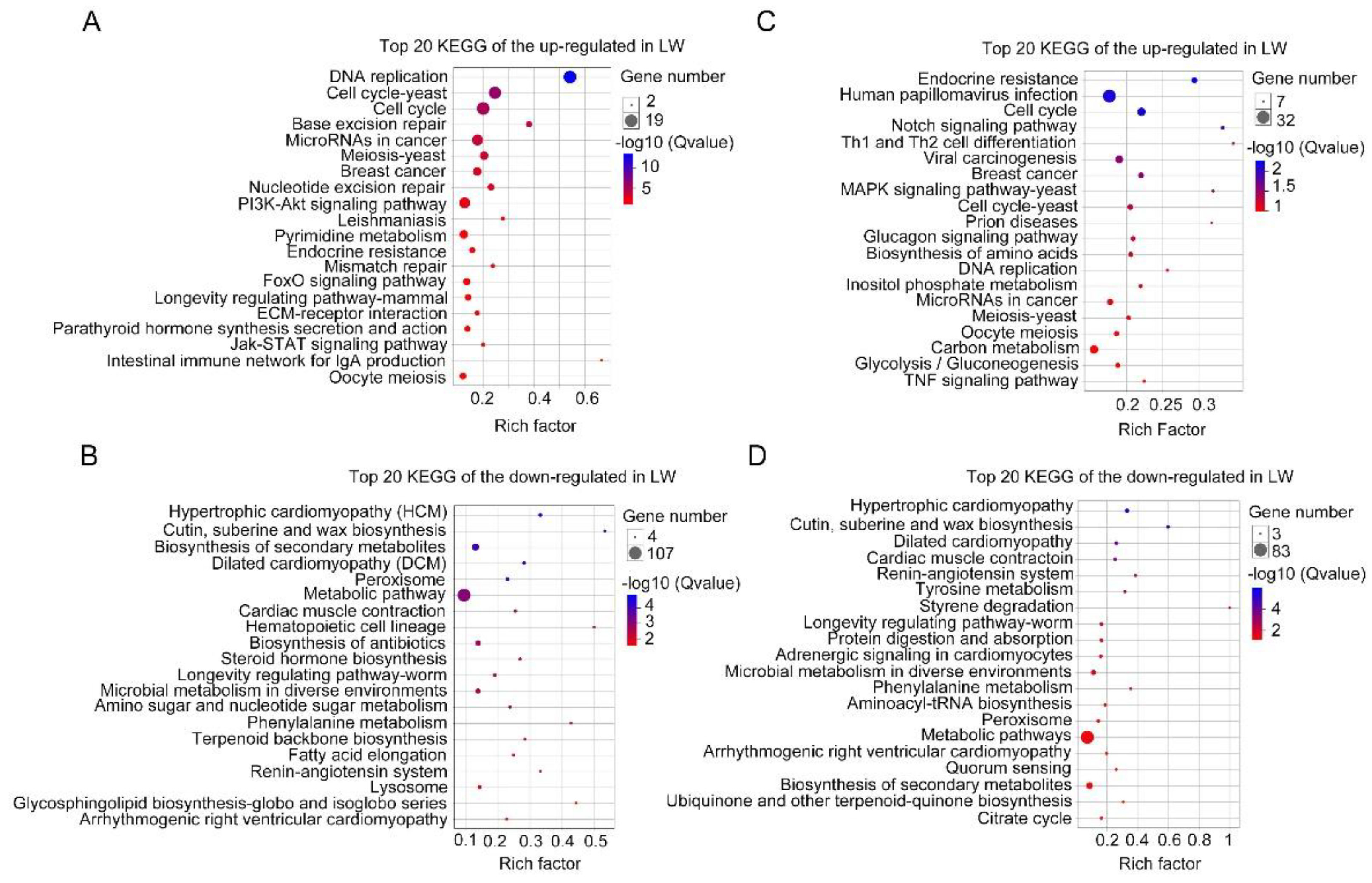
3.3. Functional Enrichment Analysis of DEGs in Forewing and Hindwing Buds at 24 hAE
3.4. Functional Enrichment Analysis of DEGs in Forewing and Hindwing Buds at 36 hAE
3.5. Functional Enrichment Analysis of DEGs in Forewing Buds and Hindwing Buds at 48 hAE
3.6. Expression Pattern of Cell Proliferation-Associated Genes in LW- and SW-Destined Wing Buds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Xue, J.; Zhou, X.; Zhang, C.X.; Yu, L.L.; Fan, H.W.; Wang, Z.; Xu, H.J.; Xi, Y.; Zhu, Z.R.; Zhou, W.W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef]
- Backus, E.A.; Serrano, M.S.; Ranger, C.M. Mechanisms of hopperburn: An overview of insect taxonomy, behavior, and physiology. Annu. Rev. Entomol. 2005, 50, 125–151. [Google Scholar] [CrossRef]
- Otuka, A. Migration of rice planthoppers and their vectored re-emerging and novel rice viruses in East Asia. Front. Microbiol. 2013, 4, 309. [Google Scholar] [CrossRef]
- Xu, H.J.; Zhang, C.X. Insulin receptors and wing dimorphism in rice planthoppers. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20150489. [Google Scholar] [CrossRef]
- Xu, H.J.; Xue, J.; Lu, B.; Zhang, X.C.; Zhuo, J.C.; He, S.F.; Ma, X.F.; Jiang, Y.Q.; Fan, H.W.; Xu, J.Y.; et al. Two insulin receptors determine alternative wing morphs in planthoppers. Nature 2015, 519, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Brisson, J.A.; Xu, H.J. Molecular Mechanisms of Wing Polymorphism in Insects. Annu. Rev. Entomol. 2019, 64, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Fu, S.J.; Chen, S.J.; Chen, H.H.; Liu, Y.L.; Liu, X.Y.; Xu, H.J. Vestigial mediates the effect of insulin signaling pathway on wing-morph switching in planthoppers. PLoS Genet. 2021, 17, e1009312. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, K.H.; Knochel, W.; Martinez, D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000, 14, 142–146. [Google Scholar] [PubMed]
- Borkhardt, A.; Repp, R.; Haas, O.A.; Leis, T.; Harbott, J.; Kreuder, J.; Hammermann, J.; Henn, T.; Lampert, F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with at(X;11) (q13; q23). Oncogene 1997, 14, 195–202. [Google Scholar] [CrossRef]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef]
- Albert, P.S.; Brown, S.J.; Riddle, D.L. Sensory control of dauer larva formation in Caenorhabditis elegans. J. Comp. Neurol. 1981, 198, 435–451. [Google Scholar] [CrossRef]
- Henderson, S.T.; Johnson, T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001, 11, 1975–1980. [Google Scholar] [CrossRef]
- Kenyon, C. A pathway that links reproductive status to lifespan in Caenorhabditis elegans. Ann. N. Y. Acad. Sci. 2010, 1204, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; Hu, P.J. Insulin/Insulin-Like Growth Factor Signaling in C. elegans; The C. elegans Research Community, Ed.; WormBook: Pasadena, CA, USA, 2013; pp. 1–43. [Google Scholar] [CrossRef]
- Hwangbo, D.S.; Gershman, B.; Tu, M.P.; Palmer, M.; Tatar, M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004, 429, 562–566. [Google Scholar] [CrossRef]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [CrossRef]
- Gross, D.N.; van den Heuvel, A.P.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef] [PubMed]
- Kousteni, S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone 2012, 50, 437–443. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; Medema, R.H.; Glassford, J.; Essers, M.A.G.; Dijkers, P.F.; Coffer, P.J.; Lam, E.W.F.; Burgering, B.M.T. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 2002, 22, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Jünger, M.A.; Rintelen, F.; Stocker, H.; Wasserman, J.D.; Végh, M.; Radimerski, T.; Greenberg, M.E.; Hafen, E. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2003, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Puig, O.; Marr, M.T.; Ruhf, M.L.; Tjian, R. Control of cell number by Drosophila FOXO: Downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003, 17, 2006–2020. [Google Scholar] [CrossRef]
- Kramer, J.M.; Davidge, J.T.; Lockyer, J.M.; Staveley, B.E. Expression of Drosophila FOXO regulates growth and can phenocopy starvation. BMC Dev. Biol. 2003, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Bao, Y.Y.; Li, B.L.; Cheng, Y.B.; Peng, Z.Y.; Liu, H.; Xu, H.J.; Zhu, Z.R.; Lou, Y.G.; Chen, J.A.; et al. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PLoS ONE 2010, 5, e14233. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, X.; Xu, X.; Li, Z.; Li, Y.; Song, D.; Yu, T.; Zhu, F.; Zhang, Q.; Zhou, X. Gene expression profiling in winged and wingless cotton aphids, Aphis gossypii (Hemiptera: Aphididae). Int. J. Biol Sci. 2014, 10, 257–267. [Google Scholar] [CrossRef]
- Shang, F.; Ding, B.Y.; Xiong, Y.; Dou, W.; Wei, D.; Jiang, H.B.; Wei, D.D.; Wang, J.J. Differential expression of genes in the alate and apterous morphs of the brown citrus aphid, Toxoptera citricida. Sci. Rep. 2016, 6, 32099. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ye, Y.X.; Zhang, H.H.; Li, D.T.; Zhuo, J.C.; Shen, Y.; Hu, Q.L.; Zhang, C.X. Chromosome-level assembly of the brown planthopper genome with a characterized Y chromosome. Mol. Ecol. Resour. 2021, 21, 1287–1298. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ziliotto, F.; Begheldo, M.; Rasori, A.; Bonghi, C.; Tonutti, P. Transcriptome profiling of ripening nectarine (Prunus persica L. Batsch) fruit treated with 1-MCP. J. Exp. Bot. 2008, 59, 2781–2791. [Google Scholar]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36 (Suppl. 1), D480–D484. [Google Scholar] [CrossRef]
- Lin, X.; Gao, H.; Xu, Y.; Zhang, Y.; Li, Y.; Lavine, M.D.; Lavine, L.C. Cell Cycle Progression Determines Wing Morph in the Polyphenic Insect Nilaparvata lugens. iScience 2020, 23, 101040. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.; Kops, G.J. Cell cycle and death control: Long live Forkheads. Trends. Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Nakamura, N.; Sansal, I.; Bergeron, L.; Sellers, W.R. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002, 2, 81–91. [Google Scholar] [CrossRef]
- Martínez-Gac, L.; Marqués, M.; García, Z.; Campanero, M.R.; Carrera, A.C. Control of cyclin G2 mRNA expression by forkhead transcription factors: Novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol. Cell Biol. 2004, 24, 2181–2189. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Yoshida-Araki, K.; Ohta, T.; Ikeda, K.; Motoyama, N. FOXO forkhead transcription factors induce G2-M checkpoint in response to oxidative stress. J. Biol. Chem. 2002, 277, 26729–26732. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, N.; Wei, S.-F.; Xu, H.-J. Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insects 2021, 12, 413. https://doi.org/10.3390/insects12050413
Xu N, Wei S-F, Xu H-J. Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insects. 2021; 12(5):413. https://doi.org/10.3390/insects12050413
Chicago/Turabian StyleXu, Nan, Sheng-Fei Wei, and Hai-Jun Xu. 2021. "Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae)" Insects 12, no. 5: 413. https://doi.org/10.3390/insects12050413
APA StyleXu, N., Wei, S.-F., & Xu, H.-J. (2021). Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insects, 12(5), 413. https://doi.org/10.3390/insects12050413