A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Termite Collection and Species Identification
2.2. Wood Treatment and Termite Feeding
2.3. Termite Dissection and DNA Isolation
2.4. 16S Amplification and Library Preparation
2.5. Sequencing, Data Processing, and Analysis
3. Results
3.1. Termite Species Identification and Feeding Bioassay
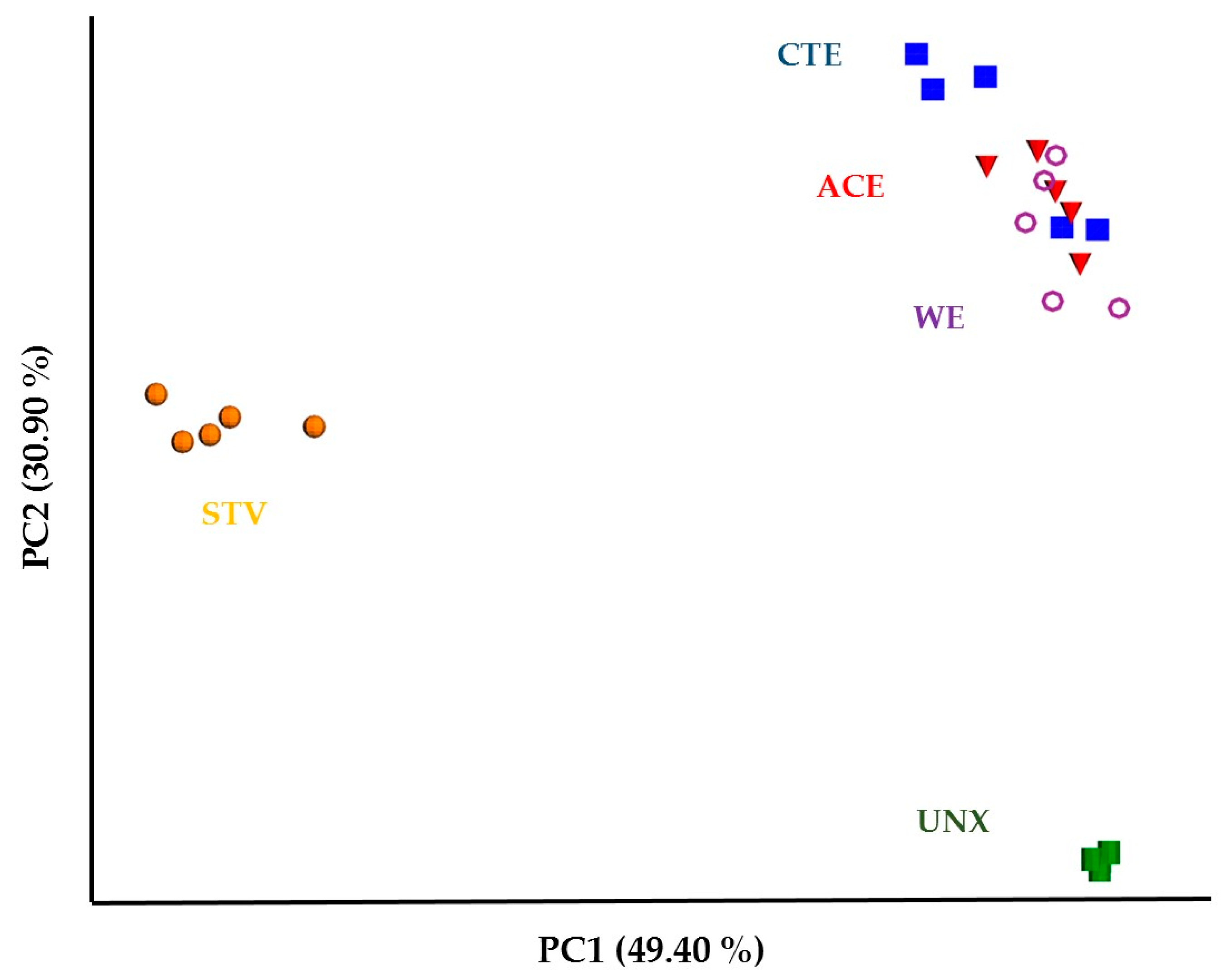
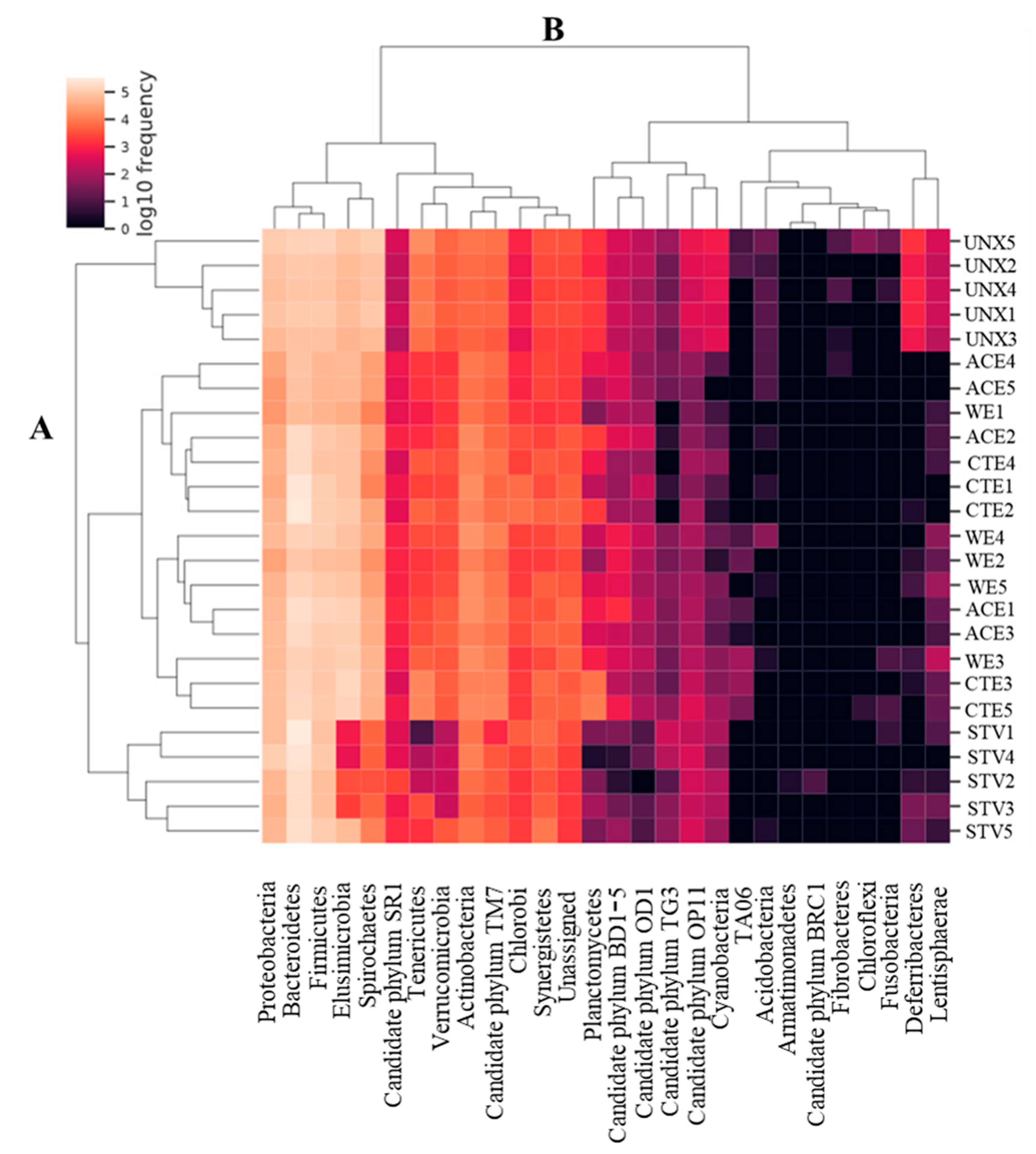
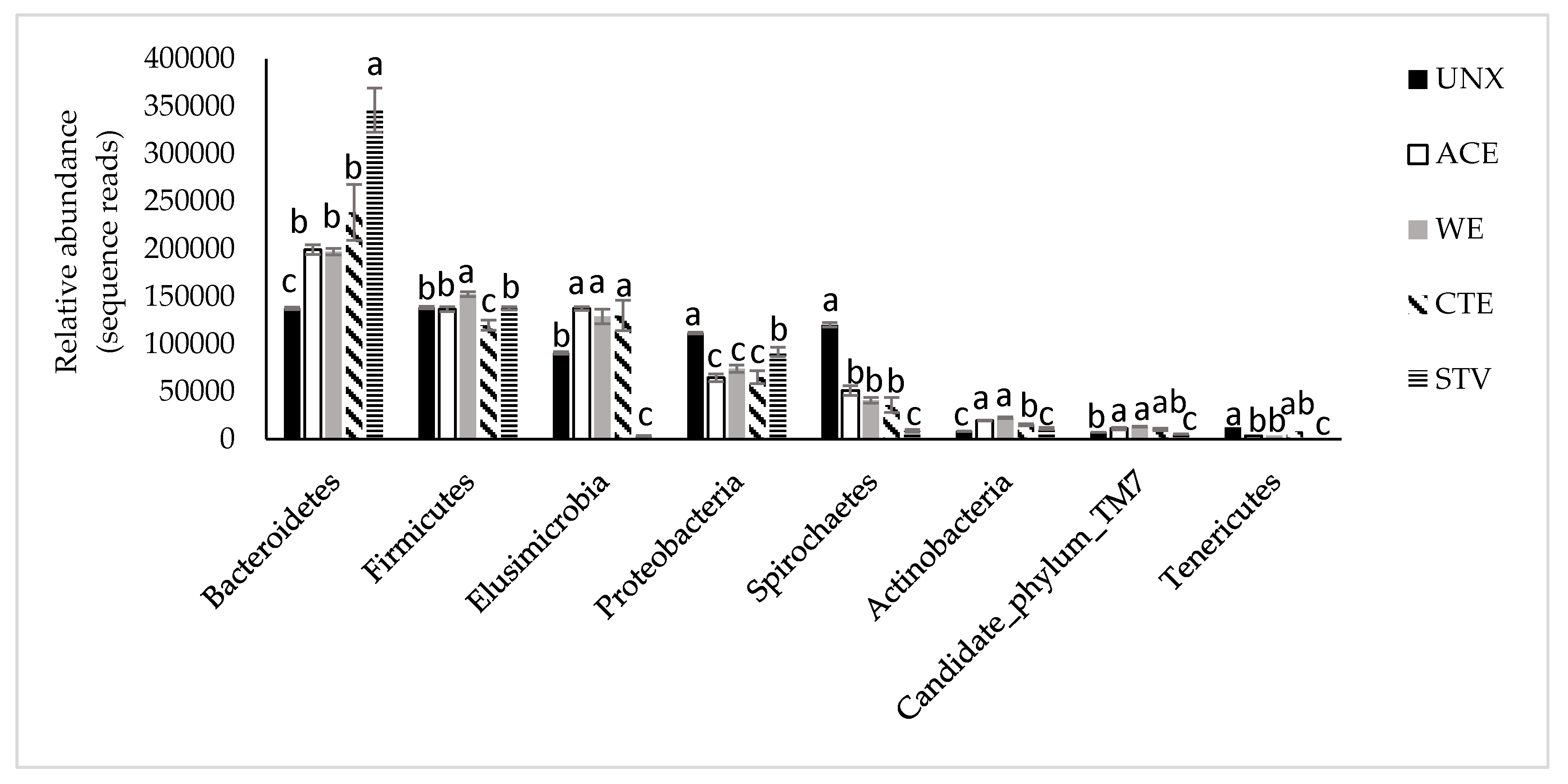
3.2. Treatment Effects on Hindgut Bacterial Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, N.Y.; Scheffrahn, R.H. Economically important termites in the United States and their control. Sociobiology 1990, 17, 77–94. [Google Scholar]
- Wang, C.; Powell, J. Survey of termites in the Delta experimental forest of Mississippi. Fla. Entomol. 2001, 84, 222–226. [Google Scholar] [CrossRef]
- Brune, A.; Dietrich, C. The gut microbiota of termites: Digesting the diversity in the light of ecology and evolution. Annu. Rev. Microbiol. 2015, 69, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Hongoh, Y. Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci. Biotechnol. Biochem. 2010, 74, 1145–1151. [Google Scholar] [CrossRef]
- Odelson, D.A.; Breznak, J.A. Volatile fatty acid production by the hindgut microbiota of xylophagous termites. Appl. Environ. Microbiol. 1983, 45, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Potrikus, C.J.; Breznak, J.A. Nitrogen-fixing Enterobacter agglomerans isolated from guts of wood-eating termites. Appl. Environ. Microbiol. 1977, 33, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Brune, A. Termite guts: The world’s smallest bioreactors. Trends Biotechnol. 1998, 16, 16–21. [Google Scholar] [CrossRef]
- Brune, A. Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 2014, 12, 168–180. [Google Scholar] [CrossRef]
- Nakajima, H.; Hongoh, Y.; Usami, R.; Kudo, T.; Ohkuma, M. Spatial distribution of bacterial phylotypes in the gut of the termite Reticulitermes speratus and the bacterial community colonizing the gut epithelium. FEMS Microbiol. Ecol. 2005, 54, 247–255. [Google Scholar] [CrossRef]
- Fisher, M.; Miller, D.; Brewster, C.; Husseneder, C.; Dickerman, A. Diversity of gut bacteria of Reticulitermes flavipes as examined by 16S rRNA gene sequencing and amplified rDNA restriction analysis. Curr. Microbiol. 2007, 55, 254–259. [Google Scholar] [CrossRef]
- Ohkuma, M.; Kudo, T. Phylogenetic diversity of the intestinal bacterial community in the termite Reticulitermes speratus. Appl. Environ. Microbiol. 1996, 62, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Hongoh, Y.; Ohkuma, M.; Kudo, T. Molecular analysis of bacterial microbiota in the gut of the termite Reticulitermes speratus (Isoptera; Rhinotermitidae). FEMS Microbiol. Ecol. 2003, 44, 231–242. [Google Scholar] [CrossRef]
- Boucias, D.G.; Cai, Y.; Sun, Y.; Lietze, V.U.; Sen, R.; Raychoudhury, R.; Scharf, M.E. The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Mol. Ecol. 2013, 22, 1836–1853. [Google Scholar] [CrossRef] [PubMed]
- Waidele, L.; Korb, J.; Voolstra, C.R.; Künzel, S.; Dedeine, F.; Staubach, F. Differential ecological specificity of protist and bacterial microbiomes across a set of termite species. Front. Microbiol. 2017, 8, 2518. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Bakker, M.G.; Judd, T.M.; Reardon, K.F.; Vivanco, J.M. Variations in diversity and richness of gut bacterial communities of termites (Reticulitermes flavipes) fed with grassy and woody plant substrates. Microb. Ecol. 2013, 65, 531–536. [Google Scholar] [CrossRef]
- Benjamino, J.; Lincoln, S.; Srivastava, R.; Graf, J. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome 2018, 6, 86. [Google Scholar] [CrossRef]
- Tharanathan, R.N.; Kittur, F.S. Chitin—the undisputed biomolecule of great potential. Crit. Rev. Food Sci. Nutr. 2003, 43, 61–87. [Google Scholar] [CrossRef]
- Raafat, D.; Sahl, H.G. Chitosan and its antimicrobial potential-a critical literature survey. Microb. Biotechnol. 2009, 2, 186–201. [Google Scholar] [CrossRef]
- Eikenes, M.; Alfredsen, G.; Christensen, B.; Militz, H.; Solheim, H. Comparison of chitosan with different molecular weights as possible wood preservative. J. Wood Sci. 2005, 51, 387–394. [Google Scholar] [CrossRef]
- Takahashi, T.; Imai, M.; Suzuki, I.; Sawai, J. Growth inhibitory effect on bacteria of chitosan membranes regulated with deacetylation degree. Biochem. Eng. J. 2008, 40, 485–491. [Google Scholar] [CrossRef]
- Goy, R.C.; Britto, D.D.; Assis, O.B.G. A review of the antimicrobial activity of chitosan. Polimeros 2009, 19, 241–247. [Google Scholar] [CrossRef]
- No, H.K.; Park, N.Y.; Lee, S.H.; Meyers, S.P. Antibacterial activity of chitosans and chitosan oligomers with different molecular weights. Int. J. Food Microbiol. 2002, 74, 65–72. [Google Scholar] [CrossRef]
- Tarras-Wahlberg, N.H.; Flachier, A.; Lane, S.N.; Sangfors, O. Environmental impacts and metal exposure of aquatic ecosystems in rivers contaminated by small scale gold mining: The Puyango River basin, southern Ecuador. Sci. Total Environ. 2001, 278, 239–261. [Google Scholar] [CrossRef]
- Liibert, L.; Treu, A.; Meier, P. The fixation of new alternative wood protection systems by means of oil treatment. J. Mater. Sci. 2011, 17, 402–406. [Google Scholar] [CrossRef]
- Raji, O.; Tang, J.D.; Telmadarrehei, T.; Jeremic, D. Termiticidal activity of chitosan against the subterranean termites Reticulitermes flavipes and Reticulitermes virginicus. Pest. Manag. Sci. 2018, 74, 1704–1710. [Google Scholar] [CrossRef]
- Telmadarrehei, T.; Tang, D.J.; Raji, O.; Rezazadeh, A.; Jeremic, D. Effect of chitosan on diversity and number of protists in subterranean termites. In Proceedings of the 114th Annual Meeting of the American Wood Protection, Seattle, WA, USA, 22–24 April 2018; Volume 114, pp. 49–59. [Google Scholar]
- Messenger, M.T. The Termite Species of Louisiana: An Identification Guide; New Orleans Mosquito and Termite Control Board: New Orleans, LA, USA, 2004; p. 4. [Google Scholar]
- Foster, B.T.; Cognato, A.I.; Gold, R.E. DNA-based identification of the eastern subterranean termite, Reticulitermes flavipes (Isoptera: Rhinotermitidae). J. Econ. Entomol. 2004, 97, 95–101. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, J.P.; Quast, C.; Horn, M.; Glokner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nuclei. Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving pictures of the human microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinformat. 2006, 2, 121–128. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Anderson, M.J.; Crist, T.O.; Chase, J.M.; Vellend, M.; Inouye, B.D.; Freestone, A.L.; Sanders, N.J.; Cornell, H.V.; Comita, L.S.; Davies, K.F.; et al. Navigating the multiple meanings of β diversity: A roadmap for the practicing ecologist. Ecol. Lett. 2011, 14, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Mikaelyan, A.; Köhler, T.; Lampert, N.; Rohland, J.; Boga, H.; Meuser, K.; Brune, A. Classifying the bacterial gut microbiota of termites and cockroaches: A curated phylogenetic reference database (DictDb). Syst. Appl. Microbiol. 2015, 38, 472–482. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Badri, M.; Kurtz, Z.; Muller, C.; Bonneau, R. Normalization methods for microbial abundance data strongly affect correlation estimates. bioRxiv. 2018. [Google Scholar] [CrossRef]
- Zhao, Y.; Park, R.D.; Muzzarelli, R.A. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef]
- Sandoval-Mojica, A.F.; Scharf, M.E. Gut genes associated with the peritrophic matrix in Reticulitermes flavipes (Blattodea: Rhinotermitidae): Identification and characterization. Arch. Insect Biochem. Physiol. 2016, 92, 127–142. [Google Scholar] [CrossRef]
- Iida, T.; Ohkuma, M.; Ohtoko, K.; Kudo, T. Symbiotic spirochetes in the termite hindgut: Phylogenetic identification of ectosymbiotic spirochetes of oxymonad protists. FEMS Microbiol. Ecol. 2000, 34, 17–26. [Google Scholar] [CrossRef]
- Stingl, U.; Radek, R.; Yang, H.; Brune, A. “Endomicrobia”: Cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl. Environ. Microbiol. 2005, 71, 1473–1479. [Google Scholar] [CrossRef]
- Hongoh, Y.; Sato, T.; Noda, S.; Ui, S.; Kudo, T.; Ohkuma, M. Candidatus Symbiothrix dinenymphae: Bristle-like Bacteroidales ectosymbionts of termite gut protists. Environ. Microbiol. 2007, 9, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.P.; Gottardi, B.; Stuepp, C.; Larré, A.B.; de Brum Vieira, P.; Tasca, T.; De Carli, G.A. Morphological aspects of Monocercomonas sp. and investigation on probable pseudocysts occurrence. Parasitol. Res. 2007, 101, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Boykin, M.S.; Stockert, L.; Buhse, H.E., Jr.; Smith-Somerville, H.E. Trichomitus trypanoides (Trichomonadida) from the termite Reticulitermes flavipes. II. Fine structure and identification of the cloned flagellate. Trans. Am. Microsc. Soc. 1986, 105, 223–238. [Google Scholar] [CrossRef]
- Tai, V.; James, E.R.; Nalepa, C.A.; Scheffrahn, R.H.; Perlman, S.J.; Keeling, P.J. The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Appl. Environ. Microbiol. 2015, 81, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, M.; Palau, M.; Guerrero, R. Gut microbiota dynamics and functionality in Reticulitermes grassei after a 7-day dietary shift and ciprofloxacin treatment. PLoS ONE 2018, 13, e0209789. [Google Scholar] [CrossRef] [PubMed]
- Janzow, M.P.; Judd, T.M. The termite Reticulitermes flavipes (Rhinotermitidae: Isoptera) can acquire micronutrients from soil. Environ. Entomol. 2015, 44, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Waidele, L.; Korb, J.; Voolstra, C.R.; Dedeine, F.; Staubach, F. Ecological specificity of the metagenome in a set of lower termite species supports contribution of the microbiome to adaptation of the host. Anim. Microbiome 2019, 1, 13. [Google Scholar] [CrossRef]
- Tang, J.D.; Raji, O.; Peterson, D.G.; Jeremic-Nikolic, D. Dysbiosis: A potential novel control strategy for control of subterranean termites. In Proceedings of the 114th Annual Meeting of the American Wood Protection Association, Seattle, WA, USA, 22–24 April 2018; Volume 114, pp. 81–91. [Google Scholar]
- Peterson, B.F.; Stewart, H.L.; Scharf, M.E. Quantification of symbiotic contributions to lower termite lignocellulose digestion using antimicrobial treatments. Insect Biochem. Mol. Biol. 2015, 59, 80–88. [Google Scholar] [CrossRef]
- Raji, O.; Tang, J.D.; Telmadarrehei, T.; Jeremic, D. Analysis of hindgut bacterial phyla frequency and diversity in subterranean termites exposed to chitosan-treated wood. In Proceedings of the IRG48 Scientific Conference on Wood Protection, Ghent, Belgium, 4–8 June 2017; Volume 48, pp. 2–8. [Google Scholar]
- Su, L.; Yang, L.; Huang, S.; Li, Y.; Su, X.; Wang, F.; Bo, C.; Wang, E.T.; Song, A. Variation in the gut microbiota of termites (Tsaitermes ampliceps) against different diets. Appl. Biochem. Biotechnol. 2017, 181, 32–47. [Google Scholar] [CrossRef]
- Tokuda, G.; Mikaelyan, A.; Fukui, C.; Matsuura, Y.; Watanabe, H.; Fujishima, M.; Brune, A. Fiber-associated spirochetes are major agents of hemicellulose degradation in the hindgut of wood-feeding higher termites. Proc. Natl. Acad. Sci. USA 2018, 115, E11996–E12004. [Google Scholar] [CrossRef]
- Hu, H.; da Costa, R.R.; Pilgaard, B.; Schiøtt, M.; Lange, L.; Poulsen, M. Fungiculture in termites is associated with a mycolytic gut bacterial community. Msphere 2019, 4, e00165-19. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y. Antibacterial activity of chitin and chitosan. Food Chem. 1988, 34, 22–29. (In Japanese) [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Treatment Solution | Percent Mass Loss * ± SE |
|---|---|
| WE | 29 ± 1.77 a |
| ACE | 26 ± 1.53 a |
| CTE | 23 ± 1.90 a |
| Treatment | Shannon’s Index ± SE | Observed OTUs ± SE | Pielou’s Evenness ± SE |
|---|---|---|---|
| UNX | 8.16 ± 0.02 a | 1823 ± 63 a | 0.75 ± 0.00 a |
| WE | 6.76 ± 0.08 b | 928 ± 82 b | 0.69 ± 0.01 b |
| ACE | 6.67 ± 0.05 b | 900 ± 51 b | 0.68 ± 0.01 b |
| CTE | 6.43 ± 0.12 b | 889 ± 61 b | 0.66 ± 0.01 b |
| STV | 6.45 ± 0.15 b | 798 ± 41 b | 0.67 ± 0.01 b |
| Method Name | PERMANOVA |
|---|---|
| Test statistic name | pseudo-F |
| Sample size | 25 |
| Number of groups | 5 |
| Test statistic | 24.1616 |
| p-value | 0.001 |
| Number of permutations | 999 |
| Pairwise treatment groups * | UNX a WE b ACE c CTE c STV d |
| Order No. | Phylum; Class; Order; Family; Genus |
|---|---|
| Bacteria found in the lowest amount in CTE samples | |
| 1 | Bacteroidetes; Bacteroidia; Bacteroidales; Porphyromonadaceae_2; Unclassified |
| 2 | Candidate phylum TM7; Termite cockroach cluster; Unclassified |
| 3 | Firmicutes; Clostridia 1; Clostridiales; ubacteriaceae 1; Anaerofustis |
| 4 | Firmicutes; Clostridia 1; Clostridiales; Family XIII Incertae Sedis; Eubacterium 3 |
| 5 | Firmicutes; Clostridia 1; Clostridiales; Lachnospiraceae; Candidatus Arthromitus |
| 6 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Unclassified |
| 7 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Anaerotruncus |
| 8 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 5 |
| 9 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 7 |
| 10 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 8 |
| 11 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 9 |
| 12 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Insect cluster |
| 13 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Incertae Sedis 1 |
| 14 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Uncultured 10 |
| 15 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Uncultured 12 |
| 16 | Firmicutes; Clostridia 2; Clostridiales 1; Peptococcaceae 1; Unclassified |
| Bacteria found in the lowest amount in CTE and STV samples | |
| 17 | Actinobacteria; Actinobacteria; Actinomycetales; Termite cluster 1; Subcluster b |
| 18 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 4 |
| 19 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Gut cluster 6 |
| 20 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Termite cockroach cluster |
| 21 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Uncultured 29 |
| 22 | Firmicutes; Clostridia 2; Clostridiales 1; Peptococcaceae 2; Uncultured gut Group A |
| 23 | Tenericutes; Mollicutes; RF9; Unclassified |
| Bacteria found in the lowest amount in STV samples, followed by the second lowest amounts in CTE samples | |
| 24 | Candidate phylum BD1-5;Unclassified |
| 25 | Firmicutes; Clostridia 1; Clostridiales; Family XIII Incertae Sedis; Unclassified |
| 26 | Firmicutes; Clostridia 1; Clostridiales; Lachnospiraceae; Catabacter |
| 27 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Papillibacter |
| 28 | Firmicutes; Clostridia 1; Clostridiales; Ruminococcaceae; Termite group aaa |
| 29 | Firmicutes; Clostridia 2; Clostridiales 1; Peptococcaceae 2; Desulfosporosinus |
| 30 | Proteobacteria; Deltaproteobacteria; Desulfovibrionales; Desulfovibrionaceae; Gut cluster 1 |
| 31 | Proteobacteria; Unclassified |
| Bacteria found in the highest amount in STV samples, followed by the second highest amounts in CTE samples | |
| 32 | Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Lactococcus 1 |
| 33 | Actinobacteria; Actinobacteria; Corynebacteriales; Corynebacteriaceae; Corynebacterium 5 |
| Increase in relative abundance in CTE when compared to UNX and STV samples | |
| 34 | Bacteroidetes; Bacteroidia; Bacteroidales; Porphyromonadaceae 2; Termite cluster 3 |
| 35 | Firmicutes; Clostridia 1; Clostridiales; Family XIII Incertae Sedis; Termite cockroach cluster 1 |
| Higher abundance in CTE and STV when compared to ACE and WE | |
| 36 | Proteobacteria; Deltaproteobacteria; Rs-K70; Termite cluster III |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telmadarrehei, T.; Tang, J.D.; Raji, O.; Rezazadeh, A.; Narayanan, L.; Shmulsky, R.; Jeremic, D. A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment. Insects 2020, 11, 681. https://doi.org/10.3390/insects11100681
Telmadarrehei T, Tang JD, Raji O, Rezazadeh A, Narayanan L, Shmulsky R, Jeremic D. A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment. Insects. 2020; 11(10):681. https://doi.org/10.3390/insects11100681
Chicago/Turabian StyleTelmadarrehei, Telmah, Juliet D. Tang, Olanrewaju Raji, Amir Rezazadeh, Lakshmi Narayanan, Rubin Shmulsky, and Dragica Jeremic. 2020. "A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment" Insects 11, no. 10: 681. https://doi.org/10.3390/insects11100681
APA StyleTelmadarrehei, T., Tang, J. D., Raji, O., Rezazadeh, A., Narayanan, L., Shmulsky, R., & Jeremic, D. (2020). A Study of the Gut Bacterial Community of Reticulitermes virginicus Exposed to Chitosan Treatment. Insects, 11(10), 681. https://doi.org/10.3390/insects11100681