Phylogenomic Analyses Clarify True Species within the Butterfly Genus Speyeria despite Evidence of a Recent Adaptive Radiation


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. RADseq and de novo Assembly
2.3. Phylogenomics
2.4. Population Genomics
3. Results
3.1. RADseq
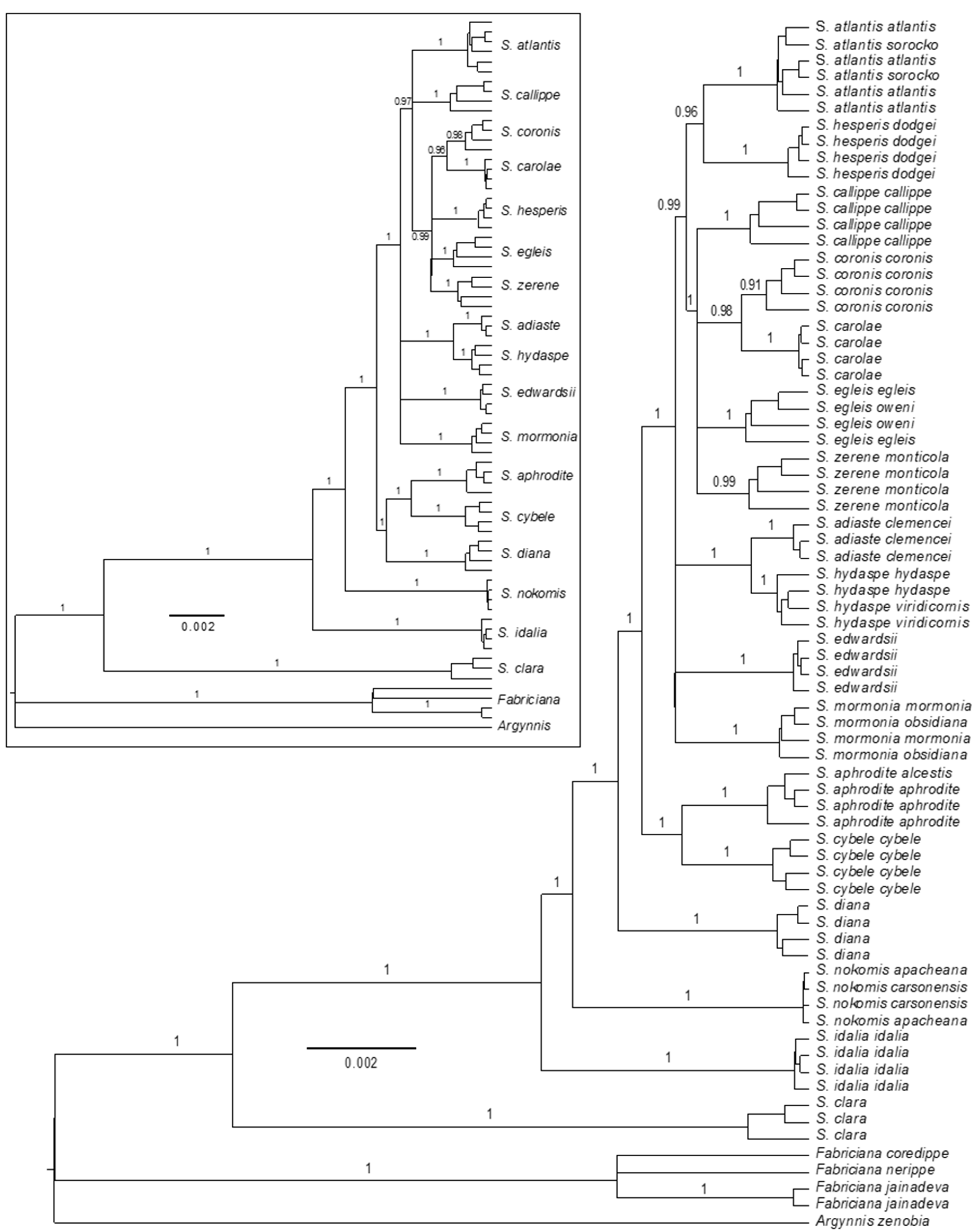
3.2. Phylogenomics
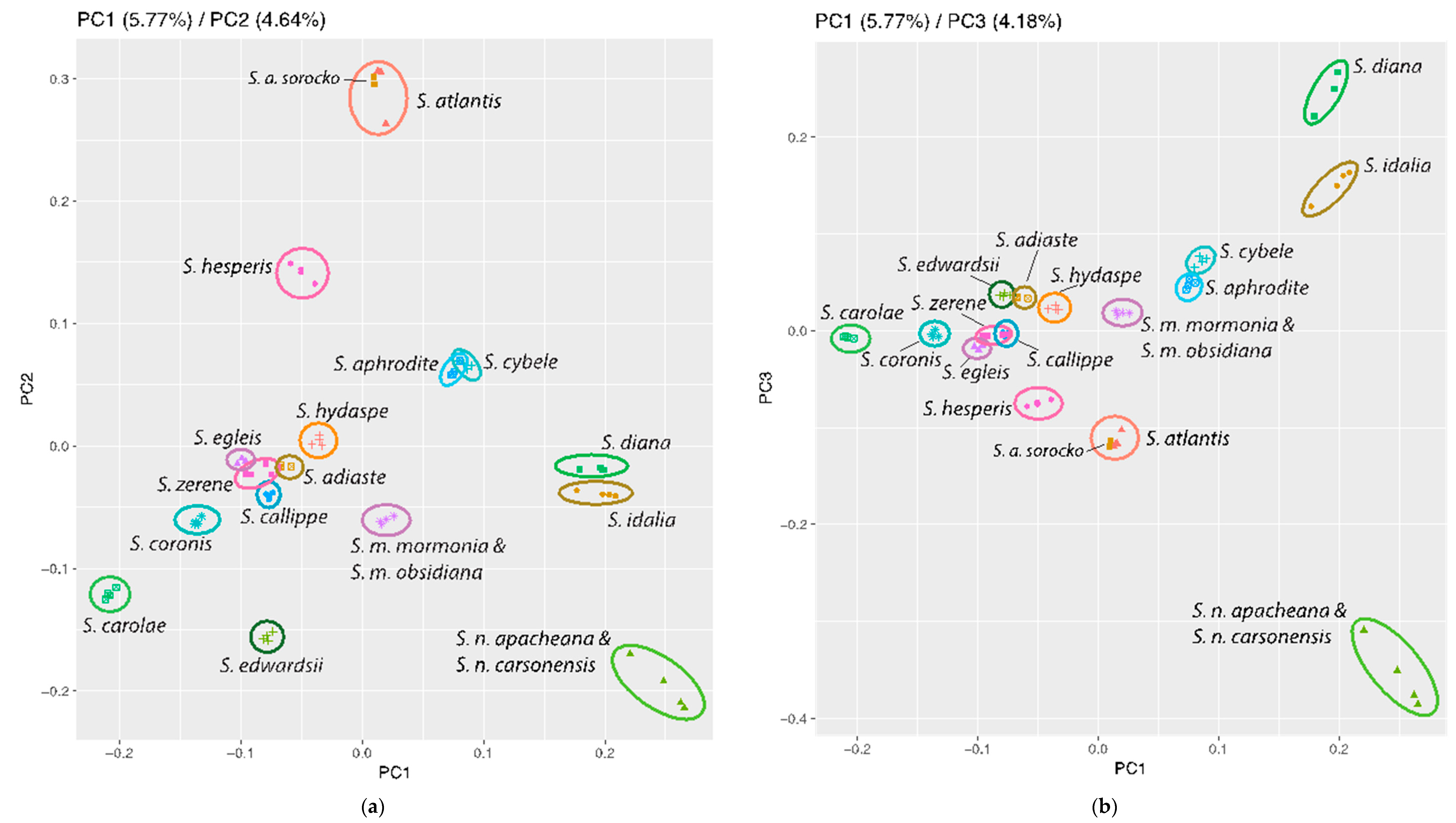
3.3. Population Genomics: PCA
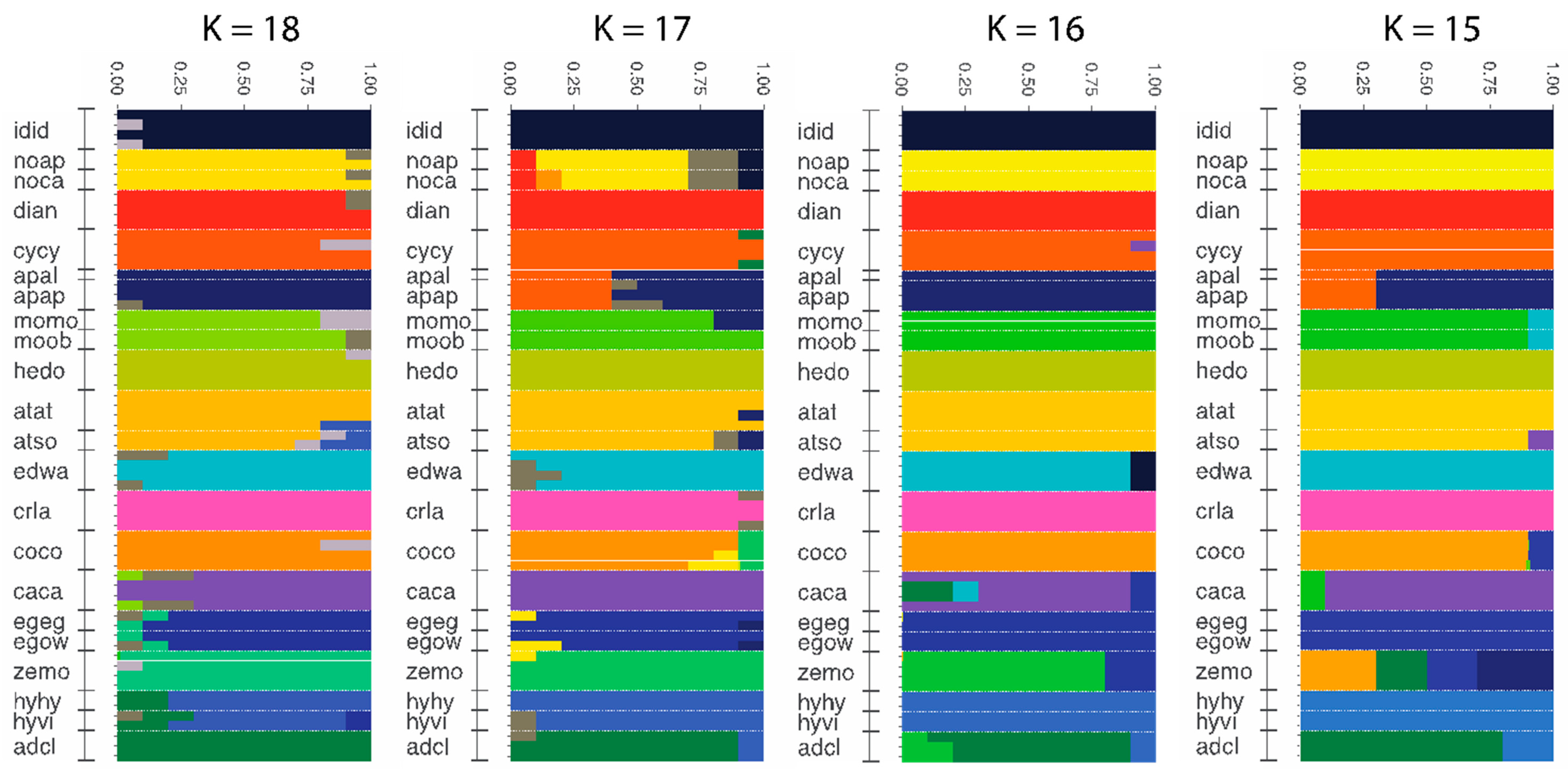
3.4. Population Genomics: Admixture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ebel, E.R.; DaCosta, J.M.; Sorenson, M.D.; Hill, R.I.; Briscoe, A.D.; Willmott, K.R.; Mullen, S.P. Rapid diversification associated with ecological specialization in Neotropical Adelpha butterflies. Mol. Ecol. 2015, 24, 2392–2405. [Google Scholar] [CrossRef] [PubMed]
- Glor, R.E. Phylogenetic insights on adaptive radiation. Annu. Rev. Ecol. Evol. Syst. 2010, 41, 251–270. [Google Scholar] [CrossRef]
- Van Bocxlaer, I.; Biju, S.; Loader, S.P.; Bossuyt, F.B. Toad radiation reveals into-India dispersal as a source of endemism in the Western Ghats-Sri Lanka biodiversity hotspot. BMC Evol. Biol. 2009, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Marshall, C.R. The true tempo of evolutionary radiation and decline revealed on the Hawaiian archipelago. Nature 2017, 543, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.D. Adaptive evolution and explosive speciation: The cichlid fish model. Nat. Rev. Genet. 2004, 5, 288–298. [Google Scholar] [CrossRef]
- Kozak, K.M.; Wahlberg, N.; Neild, A.F.E.; Dasmahapatra, K.K.; Mallet, J.; Jiggins, C.D. Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst. Biol. 2015, 64, 505–524. [Google Scholar] [CrossRef]
- Martin, S.H.; Dasmahapatra, K.K.; Nadeau, N.J.; Salazar, C.; Walters, J.R.; Simpson, F.; Blaxter, M.; Manica, A.; Mallet, J.; Jiggins, C.D. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Resour. 2013, 23, 1817–1828. [Google Scholar] [CrossRef]
- Chamberlain, N.L.; Hill, R.I.; Gilbert, L.E.; Kapan, D.D.; Kronforst, M.R. Polymorphic butterfly reveals the missing link in ecological speciation. Science 2009, 326, 847–850. [Google Scholar] [CrossRef]
- Giarla, T.C.; Esselstyn, J.A. The challenges of resolving a rapid, recent radiation: Empirical and simulated phylogenomics of philippine shrews. Syst. Biol. 2015, 64, 727–740. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Berglund, J.; Almén, M.S.; Maqbool, K.; Grabherr, M.; Martinez-Barrio, A.; Promerová, M.; Rubin, C.-J.; Wang, C.; Zamani, N.; et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 2015, 518, 371–375. [Google Scholar] [CrossRef]
- Meyer, B.S.; Matschiner, M.; Salzburger, W. A tribal level phylogeny of Lake Tanganyika cichlid fishes based on a genomic multi-marker approach. Mol. Phylogenet. Evol. 2015, 83, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Pollard, D.A.; Iyer, V.N.; Moses, A.M.; Eisen, M.B. Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genet. 2006, 2, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Sang, T.; Zhong, Y. Testing hybridization hypotheses based on incongruent gene trees. Syst. Biol. 2000, 49, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.L.; Meyer, A. Natural hybridization in primates: One evolutionary mechanism. Zoology 2006, 109, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Linder, C.R.; Rieseberg, L.H. Reconstructing patterns of reticulate evolution in plants. Am. J. Bot. 2004, 91, 1700–1708. [Google Scholar] [CrossRef] [PubMed]
- Marhold, K.; Lihová, J. Polyploidy, hybridization and reticulate evolution: Lessons from the Brassicaceae. Plant Syst. Evol. 2006, 259, 143–174. [Google Scholar] [CrossRef]
- Van Oppen, M.J.H.; Koolmees, E.M.; Veron, J.E.N. Patterns of evolution in the scleractinian coral genus Montipora (Acroporidae). Mar. Biol. 2004, 144, 9–18. [Google Scholar] [CrossRef]
- Scott, J.A. The Butterflies of North America: A Natural History and Field Guide; Stanford University Press: Stanford, CA, USA, 1986; p. 583. [Google Scholar]
- De Moya, R.S.; Savage, W.; Tenney, C.; Bao, V.; Wahlberg, N.; Hill, R.I. Interrelationships and diversification of Argynnis Fabricius and Speyeria Scudder butterflies. Syst. Entomol. 2017, 42, 635–649. [Google Scholar] [CrossRef]
- Pelham, J.P. A catalogue of the butterflies of the United States and Canada, with a complete bibliography of the descriptive and systematic literature. J. Res. Lepid. 2008, 40, 1–658. [Google Scholar]
- Dornfeld, E.J. The Butterflies of Oregon; Timber Press: Forest Grove, OR, USA, 1980. [Google Scholar]
- Warren, A.D. Butterflies of Oregon Their Taxonomy, Distribution, and Biology; Gillette Museum of Arthropod Diversity: Fort Collins, CO, USA, 2005; Volume 6. [Google Scholar]
- McHugh, A.; Bierzychudek, P.; Greever, C.; Marzulla, T.; Van Buskirk, R.; Binford, G. A molecular phylogenetic analysis of Speyeria and its implications for the management of the threatened Speyeria zerene hippolyta. J. Insect Conserv. 2013, 17, 1237–1253. [Google Scholar] [CrossRef]
- Dunford, J. Taxonomic overview of the greater fritillary genus Speyeria Scudder and the atlantis-hesperis species complexes, with species accounts, type images, and relevant literature (Lepidoptera: Nymphalidae). Insecta Mundi 2009, 0090, 1–74. [Google Scholar]
- Brittnacher, J.G.; Sims, S.R.; Ayala, F.J. Genetic differentiation between species of the genus Speyeria (Lepidoptera: Nymphalidae). Evolution 1978, 32, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, J. Butterflies through Binoculars: The West; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Hammond, P.C. The colonization of violets and Speyeria butterflies on the ash-pumice fields deposited by Cascadian volcanoes. J. Res. Lepid. 1981, 20, 179–191. [Google Scholar]
- Hammond, P.C. Patterns of geographic variation and evolution in polytypic butterflies. J. Res. Lepid. 1990, 29, 54–76. [Google Scholar]
- Hammond, P.C.; McCorkle, D.V.; Bergman, W. Hybridization studies of genomic compatibility and phenotypic expression in the Greater Fritillary Butterflies (Nymphalidae: Argynnini). J. Lepid. Soc. 2013, 67, 263–273. [Google Scholar] [CrossRef]
- Hammond, P.C.; McCorkle, D.V. The decline and extinction of Speyeria populations resulting from human environmental disturbances (Nymphalidae: Argynninae). J. Res. Lepid. 1983, 22, 217–224. [Google Scholar]
- Sims, S.R. Speyeria (Lepidoptera: Nymphalidae) Conservation. Insects 2017, 8, 45. [Google Scholar] [CrossRef]
- USFWS. U.S. Fish & Wildlife Service Endangered Species Database. Available online: http://www.fws.gov/endangered/ (accessed on 27 February 2019).
- Zaman, K.; Tenney, C.; Brunell, M.; Chen, M.; Hill, R.I. Life history and ecology of Speyeria adiaste clemencei (Comstocki, 1925) (Lepidoptera: Nymphalidae). J. Res. Lepid. 2014, 47, 29–43. [Google Scholar]
- Zaman, K.; Tenney, C.; Rush, C.E.; Hill, R.I. Population ecology of a California endemic: Speyeria adiaste clemencei. J. Insect Conserv. 2015, 19, 753–763. [Google Scholar] [CrossRef]
- Simonsen, T.J.; Wahlberg, N.; Brower, A.V.Z.; de Jong, R. Morphology, molecules and fritillaries: Approaching a stable phylogeny for Argynnini (Lepidoptera: Nymphalidae). Insect Syst. Evol. 2006, 37, 405–418. [Google Scholar]
- Simonsen, T.J. Fritillary phylogeny, classification, and larval host plants: Reconstructed mainly on the basis of male and female genitalic morphology (Lepidoptera: Nymphalidae: Argynnini). Biol. J. Linn. Soc. 2006, 89, 627–673. [Google Scholar] [CrossRef]
- Campbell, E.O.; Davis, C.S.; Dupuis, J.R.; Muirhead, K.; Sperling, F.A.H. Cross-platform compatibility of de novo-aligned SNPs in a nonmodel butterfly genus. Mol. Ecol. Resour. 2017, 17, e84–e93. [Google Scholar] [CrossRef]
- Hill, R.I.; Ganeshan, M.; Wourms, L.; Kronforst, M.R.; Mullen, S.P.; Savage, W.K. Effectiveness of DNA barcoding in Speyeria butterflies at small geographic scales. Diversity 2018, 10, 130. [Google Scholar] [CrossRef]
- Emmel, T.C.; Austin, G.T. What is Argynnis carolae? The cytotaxonomy and systematic position of a Speyeria relic from Nevada (Lepidoptera: Nymphalidae). In Systematics of Western North American Butterflies; Emmel, T.C., Ed.; Mariposa Press: Gainesville, FL, USA, 1998; pp. 443–450. [Google Scholar]
- Guppy, C.S.; Shepard, J.H. Butterflies of British Columbia; Royal British Columbia Museum and University of British Columbia Press: Victoria/Vancouver, BC, Canada, 2001; p. 414. [Google Scholar]
- Opler, P.A.; Wright, A.B. A Field Guide to Western Butterflies, 2nd ed.; Houghton Mifflin Company: New York, NY, USA, 1999. [Google Scholar]
- Pyle, R.M. The butterflies of Cascadia; Seattle Audubon Society: Seattle, WA, USA, 2002. [Google Scholar]
- Shapiro, A.M.; Manolis, T.D. Field Guide to Butterflies of the San Francisco Bay and Sacramento Valley Regions; University of California Press: Berkeley, CA, USA, 2007. [Google Scholar]
- Warren, A.D.; Davis, K.J.; Grishin, N.V.; Pelham, J.P.; Strangeland, E.M. Interactive listing of American Butterflies. 2012. [12-30-12]. Available online: http://www.butterfliesofamerica.com (accessed on 15 December 2018).
- Ali, O.A.; O’Rourke, S.M.; Amish, S.J.; Meek, M.H.; Luikart, G.; Jeffres, C.; Miller, M.R. RAD Capture (Rapture): Flexible and Efficient Sequence-Based Genotyping. Genetics 2016, 202, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Bellare, P.; DeRisi, J.L. PRICE: Software for the targeted assembly of components of (Meta) genomic sequence data. G3 Genes Genomes Genet. 2013, 3, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000_Genome_Project_Data_Processing_Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Baumsteiger, J.; Moyle, P.B.; Aguilar, A.; O’Rourke, S.M.; Miller, M.R. Genomics clarifies taxonomic boundaries in a difficult species complex. PLoS ONE 2017, 12, e0189417. [Google Scholar] [CrossRef] [PubMed]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, 1.4.4. 2017. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 September 2017).
- Fumagalli, M.; Vieira, F.G.; Linderoth, T.; Nielsen, R. ngsTools: Methods for population genetics analyses from next-generation sequencing data. Bioinformatics 2014, 30, 1486–1487. [Google Scholar] [CrossRef] [PubMed]
- R_Development_Core_Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. Pophelper: An R package and web app to analyze and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Brock, J.P.; Kaufman, K. Butterflies of North America; Houghton Mifflin Company: New York, NY, USA, 2003. [Google Scholar]
- James, D.G.; Nunnallee, D. Life Histories of Cascadia Butterflies; Oregon State University Press: Corvallis, OR, USA, 2011; p. 447. [Google Scholar]
- Britten, H.B.; Brussard, P.F.; Murphy, D.D.; Austin, G.T. Colony isolation and isozyme variability of the Western Seep Fritillary, Speyeria nokomis apacheana (Nymphalidae), in the Western Great-Basin. Great Basin Nat. 1994, 54, 97–105. [Google Scholar]
- Williams, B.L. Patterns of morphological variation in Speyeria idalia (Lepidoptera: Nymphalidae) with implications for taxonomy and conservation. Ann. Entomol. Soc. Am. 2001, 94, 239–243. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, E.; Baumsteiger, J.; Hill, R.I. Phylogenomic Analyses Clarify True Species within the Butterfly Genus Speyeria despite Evidence of a Recent Adaptive Radiation. Insects 2019, 10, 209. https://doi.org/10.3390/insects10070209
Thompson E, Baumsteiger J, Hill RI. Phylogenomic Analyses Clarify True Species within the Butterfly Genus Speyeria despite Evidence of a Recent Adaptive Radiation. Insects. 2019; 10(7):209. https://doi.org/10.3390/insects10070209
Chicago/Turabian StyleThompson, Erin, Jason Baumsteiger, and Ryan I. Hill. 2019. "Phylogenomic Analyses Clarify True Species within the Butterfly Genus Speyeria despite Evidence of a Recent Adaptive Radiation" Insects 10, no. 7: 209. https://doi.org/10.3390/insects10070209
APA StyleThompson, E., Baumsteiger, J., & Hill, R. I. (2019). Phylogenomic Analyses Clarify True Species within the Butterfly Genus Speyeria despite Evidence of a Recent Adaptive Radiation. Insects, 10(7), 209. https://doi.org/10.3390/insects10070209