Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder



Abstract
1. Introduction
2. Background
3. Morphological and Clinical Aspects of Sialidosis and NEU1 Mutation(s)
3.1. Method of Study Selection, Criteria, and Data Extraction
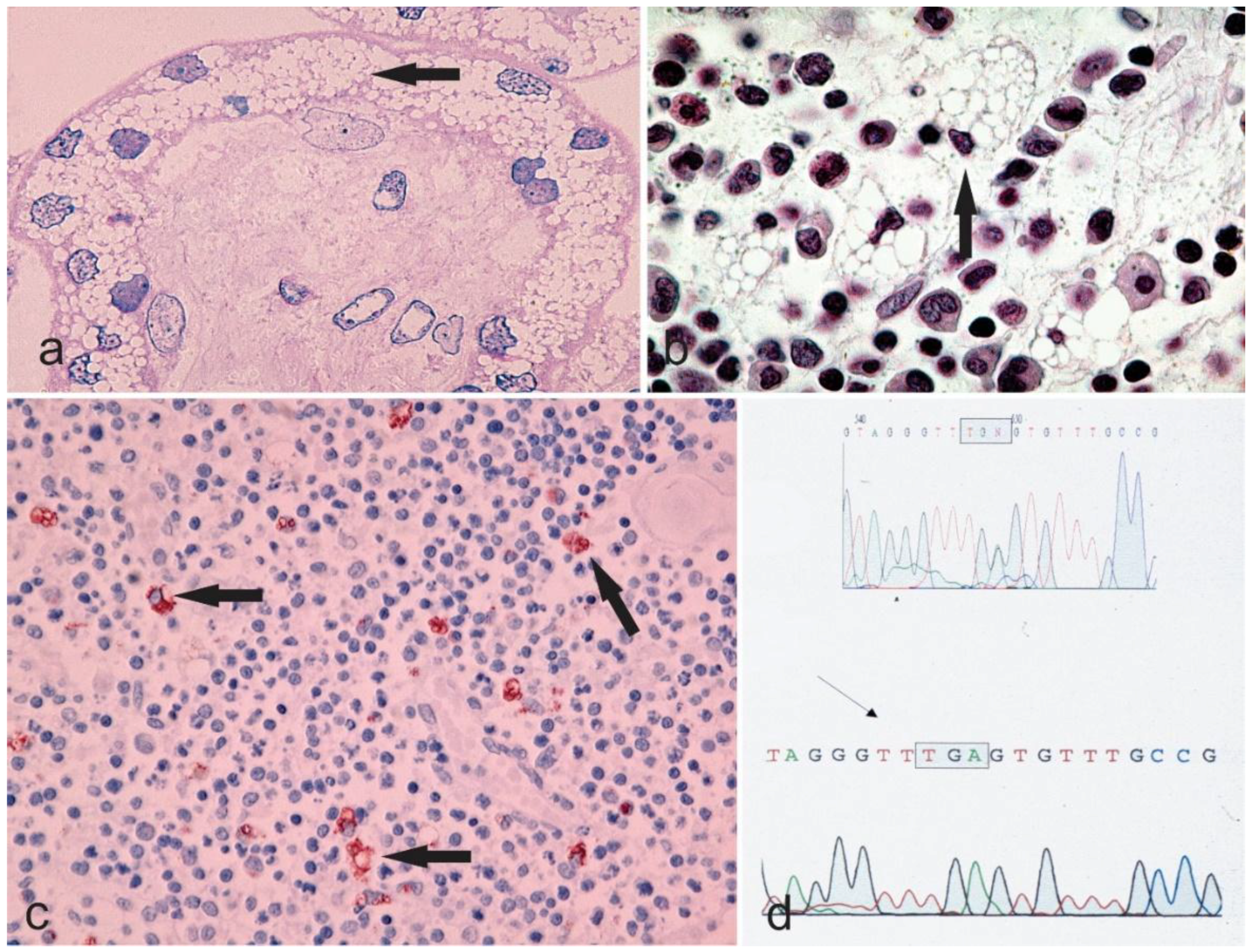
3.2. Sialidosis I
Atypical Cases of Sialidosis I
3.3. Sialidosis II
4. Therapeutic Interventions for Sialidosis
5. Conclusions and Future Perspectives
Conflicts of Interest
References
- Pshezhetsky, A.V.; Richard, C.; Michaud, L.; Igdoura, S.; Wang, S.; Elsliger, M.A.; Qu, J.; Leclerc, D.; Gravel, R.; Dallaire, L.; et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat. Genet. 1997, 15, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Cantz, M.; Gehler, J.; Spranger, J. Mucolipidosis I: Increased sialic acid content and deficiency of an alpha-n-acetylneuraminidase in cultured fibroblasts. Biochem. Biophys. Res. Commun. 1977, 74, 732–738. [Google Scholar] [CrossRef]
- Sphranger, J.; Gehler, J.; Cantz, M. Mucolipidosis i—A sialidosis. Am. J. Med. Genet. 1977, 1, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Lowden, J.A.; O’Brien, J.S. Sialidosis: A review of human neuraminidase deficiency. Am. J. Hum. Genet. 1979, 31, 1–18. [Google Scholar] [PubMed]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Arts, W.F.; Beck, M.; Covanis, A.; Donati, M.A.; Parini, R.; Zammarchi, E.; d’Azzo, A. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum. Mol. Genet. 2000, 9, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- D’Azzo, A.; Machado, E.; Annunziata, I. Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin. Orphan Drugs 2015, 3, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Elsliger, M.A.; Chang, Y.; Richard, C.; Thomas, G.; Carey, W.; Tylki-Szymanska, A.; Czartoryska, B.; Buchholz, T.; Criado, G.R.; et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum. Mol. Genet. 2000, 9, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- De Geest, N.; Bonten, E.; Mann, L.; de Sousa-Hitzler, J.; Hahn, C.; d’Azzo, A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 2002, 11, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
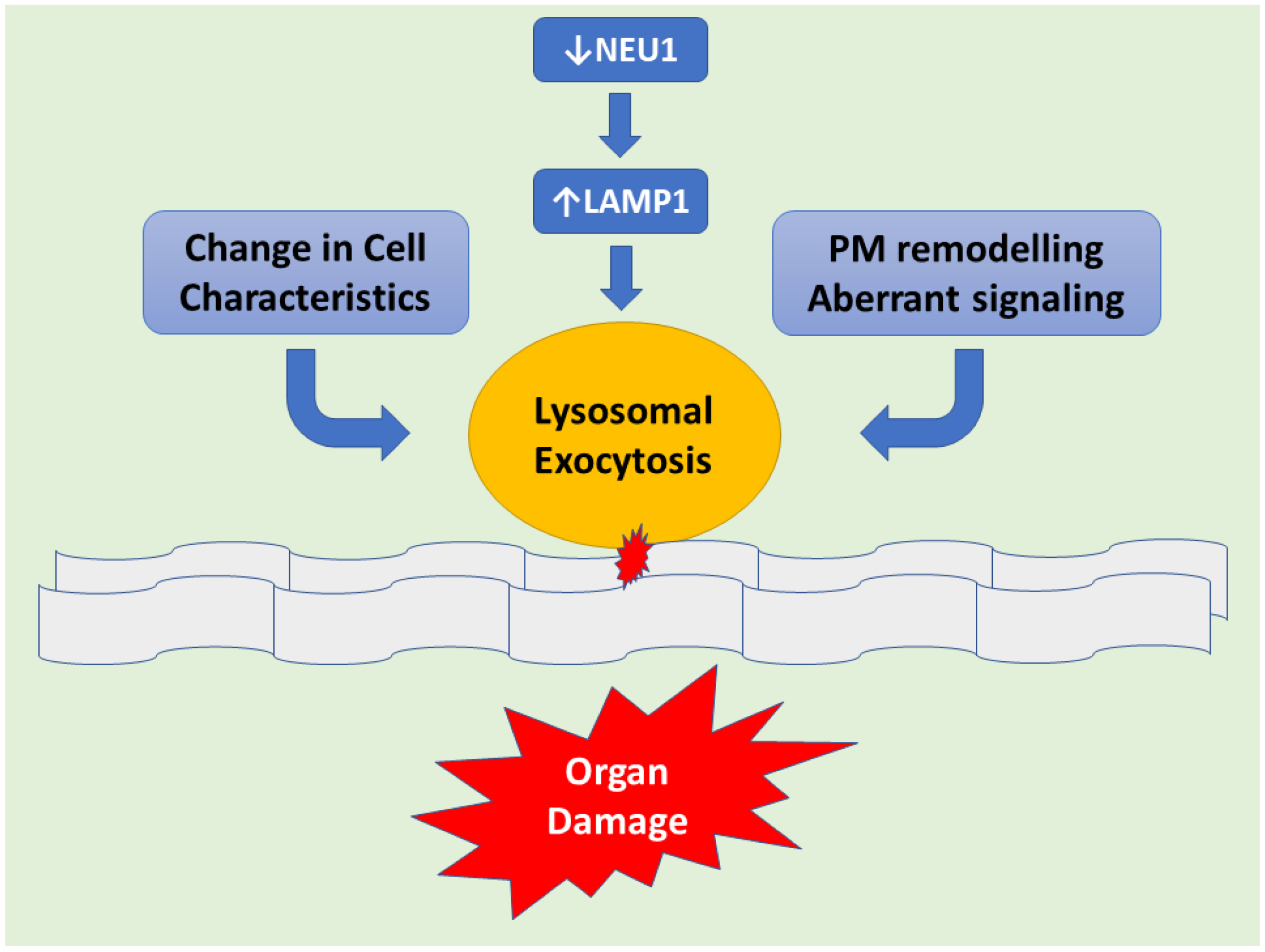
- Yogalingam, G.; Bonten, E.J.; van de Vlekkert, D.; Hu, H.; Moshiach, S.; Connell, S.A.; d’Azzo, A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 2008, 15, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Dridi, L.; Seyrantepe, V.; Fougerat, A.; Pan, X.; Bonneil, E.; Thibault, P.; Moreau, A.; Mitchell, G.A.; Heveker, N.; Cairo, C.W.; et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes 2013, 62, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Sobral, I.; Cachulo Mda, L.; Figueira, J.; Silva, R. Sialidosis type i: Ophthalmological findings. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Canafoglia, L. Sialidoses. Epileptic Disord. 2016, 18, 89–93. [Google Scholar] [PubMed]
- Takahashi, Y.; Nakamura, Y.; Yamaguchi, S.; Orii, T. Urinary oligosaccharide excretion and severity of galactosialidosis and sialidosis. Clin. Chim. Acta 1991, 203, 199–210. [Google Scholar] [CrossRef]
- Sekijima, Y.; Nakamura, K.; Kishida, D.; Narita, A.; Adachi, K.; Ohno, K.; Nanba, E.; Ikeda, S. Clinical and serial mri findings of a sialidosis type i patient with a novel missense mutation in the neu1 gene. Intern. Med. 2013, 52, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Lai, S.C.; Lu, C.S.; Weng, Y.H.; Chuang, W.L.; Chen, R.S. Abnormal cortical excitability with preserved brainstem and spinal reflexes in sialidosis type i. Clin. Neurophysiol. 2008, 119, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, S.; Villanova, M.; Malandrini, A.; van Diggelen, O.P.; Huijmans, J.G.; Ceuterick, C.; Rufa, A.; DeFalco, D.; Ciacci, G.; Martin, J.J.; et al. Type i sialidosis: A clinical, biochemical and neuroradiological study. Eur. Neurol. 2000, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.H.; Tipton, R.E.; Ch’ien, L.T.; Reynolds, L.W.; Miller, C.S. Sialidase (alpha-n-acetyl neuraminidase) deficiency: The enzyme defect in an adult with macular cherry-red spots and myoclonus without dementia. Clin. Genet. 1978, 13, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Uziel, G.; Di Donato, S.; Caimi, L.; Avanzini, G. Cherry-red spot myoclonus syndrome and alpha-neuraminidase deficiency: Neurophysiological, pharmacological and biochemical study in an adult. J. Neurol. Neurosurg. Psychiatry 1980, 43, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Canafoglia, L.; Robbiano, A.; Pareyson, D.; Panzica, F.; Nanetti, L.; Giovagnoli, A.R.; Venerando, A.; Gellera, C.; Franceschetti, S.; Zara, F. Expanding sialidosis spectrum by genome-wide screening: Neu1 mutations in adult-onset myoclonus. Neurology 2014, 82, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Di Rocco, M.; Filocamo, M.; Grossi, S.; Traverso, F.; d’Azzo, A.; Cavicchi, C.; Messeri, A.; Guerrini, R.; Zammarchi, E.; et al. Type ii sialidosis: Review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J. Neurol. 2009, 256, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.E.; Bartoshesky, L.; Harris, D.J.; McCauley, R.G.; Feingold, M.; Schott, G. Mucolipidosis i (acid neuraminidase deficiency). Three cases and delineation of the variability of the phenotype. Am. J. Dis. Child. 1981, 135, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Riches, W.G.; Smuckler, E.A. A severe infantile mucolipidosis. Clinical, biochemical, and pathologic features. Arch. Pathol. Lab. Med. 1983, 107, 147–152. [Google Scholar] [PubMed]
- Gillan, J.E.; Lowden, J.A.; Gaskin, K.; Cutz, E. Congenital ascites as a presenting sign of lysosomal storage disease. J. Pediatr. 1984, 104, 225–231. [Google Scholar] [CrossRef]
- Beck, M.; Bender, S.W.; Reiter, H.L.; Otto, W.; Bassler, R.; Dancygier, H.; Gehler, J. Neuraminidase deficiency presenting as non-immune hydrops fetalis. Eur. J. Pediatr. 1984, 143, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Guibaud, P.; Cottin, X.; Maire, I.; Boyer, S.; Guibaud, S.; Coicaud, C.; Bellon-Azzouzi, C.; Duvernois, J.P. fetal ascites as a manifestation of infantile sialidosis. Significance of a study of oligosaccharides in amniotic fluid. J. Genet. Hum. 1985, 33, 317–324. [Google Scholar] [PubMed]
- Johnson, W.G.; Thomas, G.H.; Miranda, A.F.; Driscoll, J.M.; Wigger, J.H.; Yeh, M.N.; Schwartz, R.C.; Cohen, C.S.; Berdon, W.E.; Koenigsberger, M.R. Congenital sialidosis: Biochemical studies: Clinical spectrum in four sibs; two successful prenatal diagnoses (abstract). Am. J. Hum. Genet. 1980, 32, A43. [Google Scholar]
- Yamano, T.; Shimada, M.; Matsuzaki, K.; Matsumoto, Y.; Yoshihara, W.; Okada, S.; Inui, K.; Yutaka, T.; Yabuuchi, H. Pathological study on a severe sialidosis (alpha-neuraminidase deficiency). Acta Neuropathol. 1986, 71, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Tabardel, Y.; Soyeur, D.; Vivario, E.; Senterre, J. primary neuraminidase deficiency with prenatal disclosure. Arch. Fr. Pediatr. 1989, 46, 737–740. [Google Scholar] [PubMed]
- Ries, M.; Deeg, K.H.; Wolfel, D.; Ibel, H.; Maier, B.; Buheitel, G. Colour doppler imaging of intracranial vasculopathy in severe infantile sialidosis. Pediatr. Radiol. 1992, 22, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Takahashi, Y.; Yamaguchi, S.; Omiya, S.; Orii, T.; Yara, A.; Gushiken, M. Severe infantile sialidosis—The characteristics of oligosaccharides isolated from the urine and the abdominal ascites. Tohoku J. Exp. Med. 1992, 166, 407–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmidt, M.; Fahnenstich, H.; Haverkamp, F.; Platz, H.; Hansmann, M.; Bartmann, P. sialidosis and galactosialidosis as the cause of non-immunologic hydrops fetalis. Z Geburtshilfe Neonatol. 1997, 201, 177–180. [Google Scholar] [PubMed]
- Ovali, F.; Samanci, N.; Guray, A.; Akdogan, Z.; Akdeniz, C.; Dagoglu, T.; Petorak, I. Congenital sialidosis. Turk. J. Pediatr. 1998, 40, 447–451. [Google Scholar] [PubMed]
- Sergi, C.; Beedgen, B.; Kopitz, J.; Zilow, E.; Zoubaa, S.; Otto, H.F.; Cantz, M.; Linderkamp, O. Refractory congenital ascites as a manifestation of neonatal sialidosis: Clinical, biochemical and morphological studies in a newborn syrian male infant. Am. J. Perinatol. 1999, 16, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sergi, C.; Penzel, R.; Uhl, J.; Zoubaa, S.; Dietrich, H.; Decker, N.; Rieger, P.; Kopitz, J.; Otto, H.F.; Kiessling, M.; et al. Prenatal diagnosis and fetal pathology in a turkish family harboring a novel nonsense mutation in the lysosomal alpha-n-acetyl-neuraminidase (sialidase) gene. Hum. Genet. 2001, 109, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.; Molitor, G.; Lukong, K.E.; Praun, M.; Genzel-Boroviczeny, O.; Freund, M.; Pshezhetsky, A.V.; Schulze, A. Clinical presentation of congenital sialidosis in a patient with a neuraminidase gene frameshift mutation. Eur. J. Pediatr. 2001, 160, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Uhl, J.; Penzel, R.; Sergi, C.; Kopitz, J.; Otto, H.F.; Cantz, M. Identification of a ctl4/neu1 fusion transcript in a sialidosis patient. FEBS Lett. 2002, 521, 19–23. [Google Scholar] [CrossRef]
- Donati, M.A.; Caciotti, A.; Bardelli, T.; Dani, C.; d’Azzo, A.; Morrone, A.; Zammarchi, E. Congenital sialidosis hydrops fetalis neuraminidase hydrocephalus sialidosis congenita idrope fetale neuraminidase idrocefalo. Ital. J. Pediatr. 2003, 29, 404–410. [Google Scholar]
- Penzel, R.; Uhl, J.; Kopitz, J.; Beck, M.; Otto, H.F.; Cantz, M. Splice donor site mutation in the lysosomal neuraminidase gene causing exon skipping and complete loss of enzyme activity in a sialidosis patient. FEBS Lett. 2001, 501, 135–138. [Google Scholar] [CrossRef]
- Itoh, K.; Naganawa, Y.; Matsuzawa, F.; Aikawa, S.; Doi, H.; Sasagasako, N.; Yamada, T.; Kira, J.; Kobayashi, T.; Pshezhetsky, A.V.; et al. Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes. J. Hum. Genet. 2002, 47, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Criado, G.; Pshezhetsky, A.V.; Rodriguez Becerra, A.; Gomez de Terreros, I. Clinical variability of type ii sialidosis by c808t mutation. Am. J. Med. Genet. A 2003, 116A, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Pattison, S.; Pankarican, M.; Rupar, C.A.; Graham, F.L.; Igdoura, S.A. Five novel mutations in the lysosomal sialidase gene (neu1) in type ii sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum. Mutat. 2004, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Loren, D.J.; Campos, Y.; d’Azzo, A.; Wyble, L.; Grange, D.K.; Gilbert-Barness, E.; White, F.V.; Hamvas, A. Sialidosis presenting as severe nonimmune fetal hydrops is associated with two novel mutations in lysosomal alpha-neuraminidase. J. Perinatol. 2005, 25, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Son, S.K.; Park, J.H.; Song, J.S.; Cheon, C.K. Neu1 mutation in a korean infant with type 2 sialidosis presenting as isolated fetal ascites. Pediatr. Neonatol. 2015, 56, 68–69. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, B.H.; Kim, Y.M.; Kim, J.H.; Kim, G.H.; Lee, B.S.; Kim, C.J.; Yoo, H.J.; Yoo, H.W. Histological, biochemical, and genetic characterization of early-onset fulminating sialidosis type 2 in a korean neonate with hydrops fetalis. Brain Dev. 2014, 36, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.M.; Swallow, D.M.; Baraitser, M.; Purkiss, P. Sialidosis type 2 (acid neuraminidase deficiency): Clinical and biochemical features of a further case. Clin. Genet. 1980, 18, 203–210. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Cockburn, F.; MacPhee, G.B.; Logan, R.W. Infantile type 2 sialidosis in a pakistani family—A clinical and biochemical study. J. Inherit. Metab. Dis. 1984, 7, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Oohira, T.; Nagata, N.; Akaboshi, I.; Matsuda, I.; Naito, S. The infantile form of sialidosis type ii associated with congenital adrenal hyperplasia: Possible linkage between hla and the neuraminidase deficiency gene. Hum. Genet. 1985, 70, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Young, E.P.; Mossman, J.; Fielder, A.R.; Moore, J.R. Neuraminidase deficiency: Case report and review of the phenotype. J. Med. Genet. 1987, 24, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.D.; Abeling, N.G.G.M.; Staalman, C.R.; van Gennip, A.H. Thirty-years follow-up of a patient with sialidosis. J. Inherit. Metab. Dis. 1998, 21, 116. [Google Scholar]
- Schiff, M.; Maire, I.; Bertrand, Y.; Cochat, P.; Guffon, N. Long-term follow-up of metachronous marrow-kidney transplantation in severe type ii sialidosis: What does success mean? Nephrol. Dial. Transplant. 2005, 20, 2563–2565. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Gonzalez, G.; Jimenez Lopez, I. anesthetic management of a boy with sialidosis. Rev. Esp. Anestesiol. Reanim. 2006, 53, 253–256. [Google Scholar] [PubMed]
- Ranganath, P.; Sharma, V.; Danda, S.; Nandineni, M.R.; Dalal, A.B. Novel mutations in the neuraminidase-1 (neu1) gene in two patients of sialidosis in india. Indian J. Med. Res. 2012, 136, 1048–1050. [Google Scholar] [PubMed]
- Vieira de Rezende Pinto, W.B.; Sgobbi de Souza, P.V.; Pedroso, J.L.; Barsottini, O.G. Variable phenotype and severity of sialidosis expressed in two siblings presenting with ataxia and macular cherry-red spots. J. Clin. Neurosci. 2013, 20, 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steigelman, K.A.; Bonten, E.; Hu, H.; He, W.; Ren, T.; Zuo, J.; d’Azzo, A. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim. Biophys. Acta 2010, 1802, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; d’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 2734. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Wang, D.; Toy, J.N.; Mann, L.; Mignardot, A.; Yogalingam, G.; D’Azzo, A. Targeting macrophages with baculovirus-produced lysosomal enzymes: Implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. FASEB J. 2004, 18, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bonten, E.J.; Yogalingam, G.; Mann, L.; d’Azzo, A. Short-term, high dose enzyme replacement therapy in sialidosis mice. Mol. Genet. Metab. 2005, 85, 181–189. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.M.; Igdoura, S.A. The therapeutic potential of pharmacological chaperones and proteosomal inhibitors, celastrol and mg132 in the treatment of sialidosis. Mol. Genet. Metab. 2012, 107, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Yogalingam, G.; Hu, H.; Gomero, E.; van de Vlekkert, D.; d’Azzo, A. Chaperone-mediated gene therapy with recombinant aav-ppca in a new mouse model of type i sialidosis. Biochim. Biophys. Acta 2013, 1832, 1784–1792. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| References (Name of the First Author) | Gender | General Presentation | Nervous System | Ophthalmologic Findings | Skeleton | Respiratory Distress/Infections | Renal Involvement | Cardiac Involvement | Others | Course of Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| Kelly, T.E. [23] | F | Ascites, edema, hepatosplenomegaly course features | n/r | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | n/r | Exitus at 26 months |
| Riches, W.G. [24] | F | Hydrops, ascites, hepatosplenomegaly | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 4 months |
| Gillan, J.E. [25] | M | Hydrops, ascites, edema, hepatosplenomegaly course features | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 3 days |
| Beck, M. [26] | F | Hydrops, ascites, edema, hepato-splenomegaly, coarse features | n/r | n/r | n/r | present | present | n/r | n/r | Exitus at 6 months |
| Guibaud, P. [27] | F | Hydrops, ascites, edema, hepato-splenomegaly, coarse features | n/r | Corneal clouding | Dysostosis multiplex | n/r | n/r | n/r | n/r | n/r |
| Johnson, W.G. [28] | M M F F | Hydrops, Ascites, edema | Seizures | n/r | n/r | n/r | n/r | n/r | Telangiectasia | stillborn, 1 month, 3 months, alive 3 months |
| Yamano, T. [29] | M | Hydrops, ascites, edema, hepato-splenomegaly | n/a | no | no | n/a | n/a | n/a | n/a | Exitus at 56 days |
| Tabardel, Y. [30] | n/r | Hydrops, ascites, coarse features, hepatosplenomegaly | n/r | n/r | n/r | n/r | n/r | Cardiac anomalies | Petechiae | n/r |
| Ries, M. [31] | n/a | hydrops, ascites | n/r | Cherry red spots | n/r | n/r | n/r | n/r | n/r | Exitus at 28 days |
| Lukong, K.E. [9] | F | hydrops | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 82 days |
| Nakamura, Y. [32] | F | Ascites, coarse features, hepato-splenomegaly, inguinal hernia | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | n/a | At the age of 2 months, patient was alive |
| Schmidt, M. [33] | F | Hydrops, ascites, edema, hepato-splenomegaly | Seizures | n/r | n/r | n/r | present | n/r | n/r | Exitus at 5 months |
| Ovali, F. [34] | M | Hydrops, ascites, coarse features, hepato-splenomegaly, inguinal hernia | n/r | n/r | n/r | n/r | present | n/r | n/r | Exitus at 27 days |
| Sergi, C. [35] | M | Hydrops, ascites, coarse features, hepato-splenomegaly, inguinal hernia | n/r | Corneal clouding | n/r | n/r | n/r | n/r | n/r | Exitus at 28 days |
| Sergi, C. [36] | M | Hydrops, edema ascites, hepato-splenomegaly | n/r | n/r | n/r | n/r | present | n/rn/r | n/r | Exitus at 2 months |
| Buchholz, T. [37] | M | Hydrops, edema ascites, hepato-splenomegaly | n/a | n/a | n/a | present | n/a | Cardiac anomalies | Telangiectasia Hypotonia | Exitus at 82 days |
| Uhl, J. [38] | M M | Hydrops, edema, ascites in both patients | n/a | n/a | n/a | n/a | n/a | n/r | Polydactyly in patient 1 | n/a |
| Donati, M.A. [39] | F | Hydrops, ascites, edema, coarse features, hepato-splenomegaly, Inguinal hernia | Psychomotor retardation, Hydrocephalus | yellow/rretina | Dysostosis multiplex | present | present | Cardiac anomalies | Telangiectasia Hypotonia, Petechiae | Exitus at 19 months |
| Penzel, R. [40] | F | Hydrops, ascites, edema | Seizures | n/r | n/r | n/r | n/r | n/r | n/r | n/r |
| Itoh, K. [41] | M | Hydrops, ascites, edema, hepato-splenomegaly | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 27 days |
| Rodriguez Criado, G. [42] | M | Hydrops, coarse features, hepatosplenomegaly | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | Hypotonia | Exitus at 20 months |
| Pattison, S. [43] | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at Patient 1; 3 months Patient 2; 2 months |
| Loren, D.J. [44] | n/r | Hydrops, ascites, edema, hepato-splenomegaly | n/r | n/r | no | n/r | n/r | n/r | n/r | Alive at the age of three months |
| Caciotti, A. [22] | F | Coarse features, hepatosplenomegaly | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | present | Cardiac anomalies | Telangiectasia Hypotonia, petechiae | Exitus at 1 year |
| Bonten, E.J. [7] | F | Hydrops, hepato-splenomegaly | Psychomotor retardation, Hydrocephalus | No corneal opacity | Dysostosis multiplex, Joint contractures | n/r | n/r | cardiomyopathy | Cardiac anomalies | Exitus at 18 months |
| Lee, Y.J. [45] | F | Hydrops, ascites, coarse features, hepatosplenomegaly | n/r | Bilateral congenital cataracts with foveal hypoplasia | n/r | n/r | n/r | n/r | Telangiectasia Hypotonia, bluish to purpuric macules mild thrombocytopenia | Exitus a t9 months |
| Lee, B.H. [46] | F | Hydrops, ascites, coarse features, edema, hepato-splenomegaly | n/r | n/r | n/r | Present | n/r | Cardiomegaly with huge patent ductus arteriosus (PDA), Ventriculomegaly | Hypotonia | Exitus a t3 months |
| References (Name of the First Author) | Gender | General Presentation | Nervous System | Ophthalmologic Findings | Skeleton | Respiratory Distress/Infections | Renal Involvement | Cardiac Involvement | Others | Course of Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| Winter, R.M. [47] | M <1 year | Coarse feature | Psychomotor delay, Seizures | Visual loss | Dysostosis Multiplex | n/r | n/r | n/r | Hearing loss, Inguinal hernia | 22 years |
| Kelly, T.E. [23] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, seizures/myoclonic jerks | Cherry red spot | Dysostosis Multiplex | present | n/r | present | Umbilical hernia | 5 and half years |
| Kelly, T.E. [23] | F Birth | Coarse feature, Hepatosplenomegaly | Psychomotor delay | Cataract | Dysostosis Multiplex | n/r | n/r | present | Hearing loss, Umbilical hernia, Hypotonia | 24 months |
| King, M. [48] | M 5 months | Coarse feature, Hepatosplenomegaly | Psychomotor delay Ataxia | Cherry red spot, corneal Clouding, Cataract, | Dysostosis Multiplex | n/r/ | n/r | n/r | Hearing loss | 13 years |
| King, M. [48] | F N/A | Coarse feature, Hepatosplenomegaly | Psychomotor delay | Cherry red spot, Cataract, | Dysostosis Multiplex | n/r | n/r | n/r | Hearing loss | 12 years |
| Oohira, T. [49] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, Ataxia, myoclonic jerks | Cherry red spots | Dysostosis Multiplex | n/r | n/r | n/r | Hypotonia | 5 years |
| Young, I.D. [50] | M 18 months | Coarse feature | Psychomotor delay, Ataxia, myoclonic jerks | Cherry red spot, Nystagmus, Optic atrophy | Dysostosis Multiplex | nr | n/r | n/r | Hearing Loss, Hypotonia | 12 years |
| Bakker, H.D. [51] | F 6 months | Coarse feature | Psychomotor delay | Strabismus, Nystagmus | n/r | n/r | n/r | n/r | Hearing Loss, Hypotonia | 30 years |
| Rodriguez Criado, G. [42] | M <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, Myoclonic movements Ataxia | n/r | Dysostosis Multiplex | n/r | n/r | present | Hearing Loss, Hypotonia | 13 years |
| Rodriguez Criado, G. [42] | M 16 months | Coarse feature, Hepatosplenomegaly | Psychomotor delay | n/r | Dysostosis Multiplex | n/r | n/r | absent | Hearing Loss, Hypotonia | 11 years |
| Pattison, S. [43] | n/r | Coarse feature, Hepatosplenomegaly | n/r | n/r | Dysostosis Multiplex | n/r | n/r | n/r | n/r | 3 years |
| Pattison, S. [43] | n/r | Coarse feature, Hepatosplenomegaly | n/r | n/r | Dysostosis Multiplex | n/r | n/r | n/r | n/r | 3 years |
| Schiff, M. [52] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay | n/r | Dysostosis Multiplex | n/r | present | n/r | n/r | 11 years |
| Gonzalez Gonzalez G [53] | n/r | n/r | Myoclonic epilepsy | n/r | Dysostosis Multiplex | n/r | n/r | present | n/r | 14 years |
| Caciotti, A. [22] | M 1 year | Coarse feature | Psychomotor delay Seizures | Cherry red spot, cataract, | Dysostosis Multiplex | n/r | n/r | n/r | Hearing Loss | 9 years |
| Bonten, E.J. [7] | M birth | Coarse feature, Hepatosplenomegaly | Developmental delay, Orbital hypoplasia | normal | Craniosynostosis | n/r | n/r | n/r | n/r | Progressing at 4 months |
| Bonten, E.J. [7] | F 12 years | n/r | Psychomotor delay, Seizures Ataxia, Dysmetria Spasticity | Cherry red spots | Dysostosis Multiplex, microcephaly | n/r | n/r | ECG specific alterations of repolarization | Hearing Lossl | Progressing at 28 years. |
| Ranganath, P. [54] | F 18 months | Coarse facies, hepatomegaly | n/a | Mild corneal haziness, bilateral fundal Cherry red spots | Macrocephaly | n/r | n/r | Cardiac anomalies | Protuberant tongue, gum hypertrophy, generalized hypertrichosis, large Mongolian spots on the back, umbilical hernia | n/r |
| de Rezende Pintoi ** [55] | F 30 years | a high forehead and low-set ears | Advanced degree of mental deficiency.lower limb spasticity, and facial and limb myoclonic jerks | Bilateral macular cherry-red spots | n/r | n/r | n/r | n/r | A marked delayed in motor and cognitive functions present since childhood. Cognitive and motor skills had worsened over 10 years | n/r |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Sergi, C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics 2018, 8, 29. https://doi.org/10.3390/diagnostics8020029
Khan A, Sergi C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics. 2018; 8(2):29. https://doi.org/10.3390/diagnostics8020029
Chicago/Turabian StyleKhan, Aiza, and Consolato Sergi. 2018. "Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder" Diagnostics 8, no. 2: 29. https://doi.org/10.3390/diagnostics8020029
APA StyleKhan, A., & Sergi, C. (2018). Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics, 8(2), 29. https://doi.org/10.3390/diagnostics8020029