
Clinical, Radiographic, and Molecular Analysis of Patients with X-Linked Hypophosphatemic Rickets: Looking for Phenotype–Genotype Correlation

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Molecular Test
2.3. Rickets Severity Score
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chanchlani, R.; Nemer, P.; Sinha, R.; Nemer, L.; Krishnappa, V.; Sochett, E.; Safadi, F.; Raina, R. An Overview of Rickets in Children. Kidney Int. Rep. 2020, 5, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.D.; Demay, M.B.; Burnett-Bowie, S.A.M. The biology and pathology of vitamin D control in bone. J. Cell. Biochem. 2010, 111, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Winters, R.W.; Graham, J.V.; Williams, T.F.; Mcfalls, V.W.; Burnett, C.H. A genetic study of familial hypophosphatemia and vitamin D resistant rickets with a review of the literature. Medicine 1958, 37, 97–142. [Google Scholar] [CrossRef] [PubMed]
- Padidela, R.; Nilsson, O.; Nilsson, O.; Makitie, O.; Beck-Nielsen, S.; Ariceta, G.; Schnabel, D.; Brandi, M.L.; Boot, A.; Levtchenko, E.; et al. The international X-linked hypophosphataemia (XLH) registry (NCT03193476): Rationale for and description of an international, observational study. Orphanet J. Rare Dis. 2020, 15, 172. [Google Scholar] [CrossRef]
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; De Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef]
- Bitzan, M.; Goodyer, P.R. Hypophosphatemic Rickets. Pediatr. Clin. N. Am. 2018, 66, 179–207. [Google Scholar] [CrossRef]
- Amanzadeh, J.; Reilly, R.F. Hypophosphatemia: An evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol. 2006, 2, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ooshima, T.; Lily, T.S.; Yasufuku, Y.; Sobue, S. Structural deformities of deciduous teeth in patients with hypophosphatemic vitamin D-resistant rickets. Oral Surg. Oral Med. Oral Pathol. 1988, 65, 191–198. [Google Scholar] [CrossRef]
- Chaussain-Miller, C.; Sinding, C.; Septier, D.; Wolikow, M.; Goldberg, M.; Garabedian, M. Dentin structure in familial hypophosphatemic rickets: Benefits of vitamin D and phosphate treatment. Oral Dis. 2007, 13, 482–489. [Google Scholar] [CrossRef]
- Shore, R.M.; Chesney, R.W. Rickets: Part II. Pediatr. Radiol. 2013, 43, 152–172. [Google Scholar] [CrossRef]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, A.E. The acrophysis: A unifying concept for understanding enchondral bone growth and its disorders. II. Abnormal growth. Skelet. Radiol. 2004, 33, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Cheema, J.I.; Grissom, L.E.; Harcke, H.T. Radiographic Characteristics of Lower-Extremity Bowing in Children. Radiographics 2003, 23, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Thacher, T.D.; Pettifor, J.M.; Tebben, P.J.; Creo, A.L.; Skrinar, A.; Mao, M.; Chen, C.Y.; Chang, T.; San Martin, J.; Carpenter, T.O. Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: Utility of the radiographic Rickets Severity Score. Bone 2019, 122, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; van’t Hoff, W.; Mao, M.; Chen, C.-Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, B.H.; Kang, J.H.; Ha, I.S.; I Cheong, H.; Choi, Y. A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr. Res. 2005, 58, 329–333. [Google Scholar] [CrossRef]
- Holm, I.A.; Nelson, A.E.; Robinson, B.G.; Mason, R.S.; Marsh, D.J.; Cowell, C.T.; Carpenter, T.O. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 2001, 86, 3889–3899. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, E.; Gil-Peña, H.; Chocron, S.; Madariaga, L.; de la Cerda-Ojeda, F.; Fernández-Fernández, M.; de Lucas-Collantes, C.; Gil, M.; Luis-Yanes, M.I.; Vergara, I.; et al. Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population. Orphanet J. Rare Dis. 2021, 16, 104. [Google Scholar] [CrossRef]
- Jiménez, M.; Ivanovic-Zuvic, D.; Loureiro, C.; Carvajal, C.A.; Cavada, G.; Schneider, P.; Gallardo, E.; García, C.; Gonzalez, G.; Contreras, O.; et al. Clinical and molecular characterization of Chilean patients with X-linked hypophosphatemia. Osteoporos. Int. 2021, 32, 1825–1836. [Google Scholar] [CrossRef]
- Rafaelsen, S.; Johansson, S.; Ræder, H.; Bjerknes, R. Hereditary hypophosphatemia in Norway: A retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur. J. Endocrinol. 2016, 174, 125–136. [Google Scholar] [CrossRef]
- Lin, X.; Li, S.; Zhang, Z.; Yue, H. Clinical and Genetic Characteristics of 153 Chinese Patients With X-Linked Hypophosphatemia. Front. Cell Dev. Biol. 2021, 9, 617738. [Google Scholar] [CrossRef] [PubMed]
- Park, P.G.; Lim, S.H.; Lee, H.; Ahn, Y.H.; Cheong, H.I.; Kang, H.G. Genotype and Phenotype Analysis in X-Linked Hypophosphatemia. Front. Pediatr. 2021, 9, 699767. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, C.; Walrant-Debray, O.; Nguyen, T.M.; Esterle, L.; Garabédian, M.; Jehan, F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum. Genet. 2009, 125, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Ruppe, M.D.; Brosnan, P.; Au, K.S.; Tran, P.X.; Dominguez, B.W.; Hope, N. Mutational analysis of PHEX, FGF23 and DMP1 in a cohort of patients with hypophosphatemic rickets. Clin. Endocrinol. 2011, 74, 312–318. [Google Scholar] [CrossRef]
- Živičnjak, M.; Schnabel, D.; Billing, H.; Staude, H.; Filler, G.; Querfeld, U.; Schumacher, M.; Pyper, A.; Schröder, C.; Brämswig, J.; et al. Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr. Nephrol. 2011, 26, 223–231. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, C.; Chen, Q.; Che, R.; Sha, Y.; Zhao, F.; Ding, G.; Zhou, W.; Jia, Z.; Huang, S.; et al. Functional Characterization of PHEX Gene Variants in Children With X-Linked Hypophosphatemic Rickets Shows No Evidence of Genotype–Phenotype Correlation. J. Bone Miner. Res. 2020, 35, 1718–1725. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Z.; Sun, Y.; Xu, L.; JiaJue, R.; Cui, L.; Pang, Q.; Jiang, Y.; Li, M.; Wang, O.; et al. Clinical and genetic analysis in a large Chinese cohort of patients with X-linked hypophosphatemia. Bone 2019, 121, 212–220. [Google Scholar] [CrossRef]
- Borghi, M.; da Silva, L.M.; Bispo, L.; Longui, C.A. A genetic study of a Brazilian cohort of patients with X-linked hypophosphatemia reveals no correlation between genotype and phenotype. Front. Pediatr. 2023, 11, 1215952. [Google Scholar] [CrossRef]
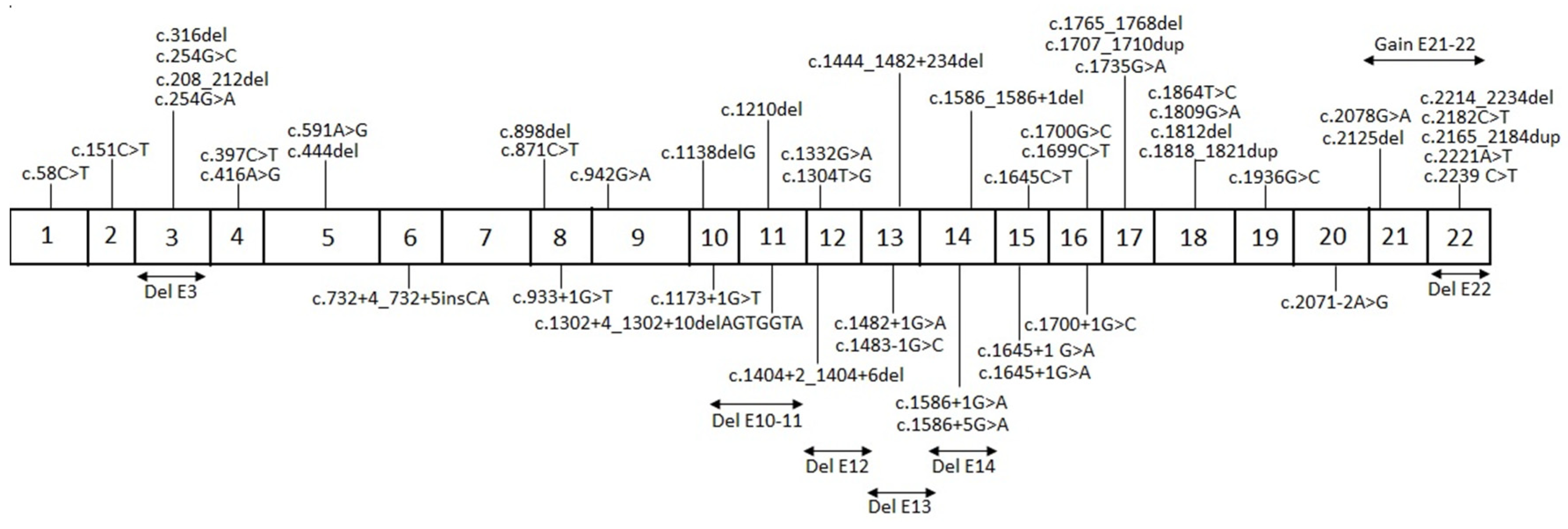
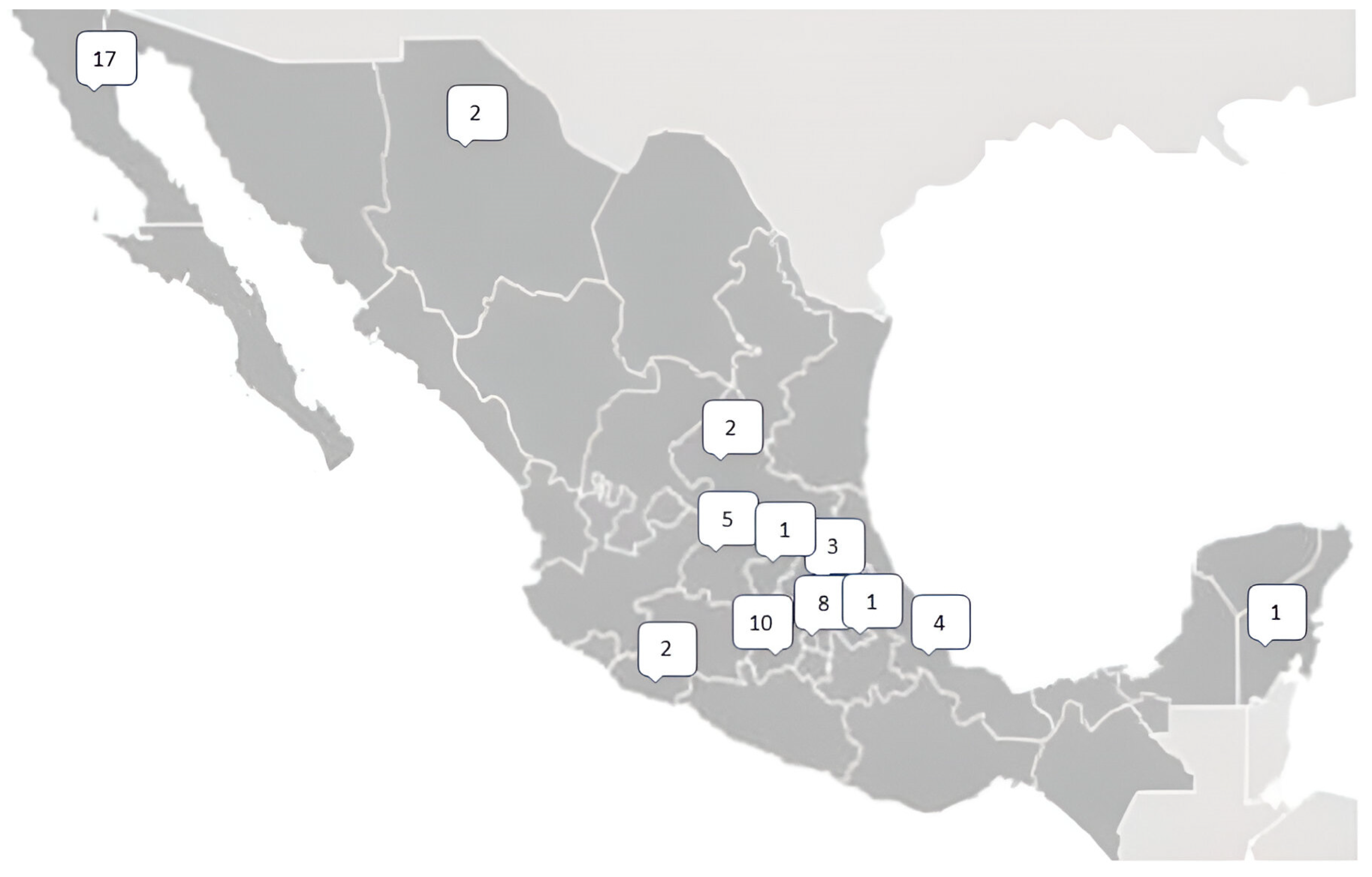



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Variant | Consequence | Age | RSS | Ca mg/dL | P mmol/L | ALP U/L |
|---|---|---|---|---|---|---|---|---|
| Px 4 | F | c.2239 C > T (p.Arg747*) | Nonsense | 10 | 1.25 | 139.4 | 3 | 455.6 |
| Px 5 | M | c.1700 + 1G > C (p.?) | Splice variant | 2 | 3 | 140.8 | 2.9 | 379.6 |
| Px 7 | M | c.1302 + 4_1302 + 10delAGTGGTA (p.?) | Splice variant | 2 | 4.5 | 139.6 | 2.9 | 394.4 |
| Px 14 | M | c.1483-1G > C (p.?) | Splice variant | 16 | 4.5 | 139.6 | 2.4 | 252.7 |
| Px 35 | F | c.1735G > A (p.Gly579Arg) | Missense | 8 | 0.5 | 136.2 | 2.4 | 800 |
| Px 37 | F | c.1645 + 1 G>A | Splice variant | 30 | 3.75 | 138.8 | 2.6 | 647.5 |
| Px 44 | F | c.1735G > A (p.Gly579Arg) | Missense | 17 | 2.5 | 10 | 2.6 | 623 |
| Px 45 | F | c.1707_1710dup (p.Tyr571Glufs*12) | Frameshift | 9 | 5.5 | 9.5 | 2.8 | 420 |
| Px 46 | F | c.1304T > G (p.Met435Arg) | Missense | 8 | 2.5 | 10.8 | 3.1 | 487 |
| Px 48 | M | Deletion exon 22 | Frameshift | 11 | 1.25 | 9.3 | 2.9 | 427 |
| Px 50 | F | c.1735G > A (p.Gly579Arg) | Missense | 13 | 2.75 | 9.3 | 2.5 | 539 |
| Px 51 | F | c.1586 + 5G>A | Splice variant | 16 | 4 | 10.1 | 2.8 | 581 |
| Px 52 | F | c.444del (p.Ile148Metfs*73) | Frameshift | 13 | 1.25 | 9.9 | 3.1 | 459 |
| Px 53 | M | c.871C > T (p.Arg291*) | Nonsense | 15 | 4.25 | 9.3 | 3.3 | 606 |
| Px 57 | M | c.1765_1768del (p.Asn589Valfs*29) | Frameshift | 6 | 4 | 9.7 | 2.9 | 557 |
| Px 59 | F | c.2221A > T (p.Arg741*) | Nonsense | 15 | 2 | 10 | 2.9 | 512 |
| Px 60 | M | c.58C > T (p.Arg20*) | Nonsense | 15 | 3 | 9.6 | 2.9 | 571 |
| Px 61 | F | Deletion (Exon 3) | Frameshift | 14 | 1.25 | 9.2 | 2.8 | 404 |
| Px 62 | F | c.1645C > T (p.Arg549*) | Nonsense | 10 | 4 | NA | NA | NA |
| Px 65 | F | c.1936G > C (p.Asp646His) | Missense | 6 | 3.25 | 10 | 3 | 837 |
| Px 66 | M | c.942G > A (p.Trp314*) | Missense | 7 | 1.75 | 9.6 | 2.9 | 355 |
| Px 69 | M | c.2125del (p.Ala709Leufs*31) | Frameshift | 20 | 2.5 | 9.9 | 3.3 | 418 |
| Px 71 | M | c.1332G > A (p.Trp444*) | Nonsense | 8 | 2.25 | 9.3 | 3.2 | 525 |
| Px 73 | F | c.1332G > A (p.Trp444*) | Nonsense | 4 | 3 | NA | NA | NA |
| Px 74 | F | c.1645 + 1G>A | Splice variant | 18 | 2.75 | 7.4 | 2 | 1155 |
| Px 76 | F | c.2165_2184dup (p.Lys729Valfs*18) | Frameshift | 16 | 5 | 8.5 | 2.5 | 1363 |
| Px 78 | M | c.2182C > T (p.Gln728*) | Nonsense | 10 | 6 | 10 | 3.5 | 754 |
| Px 80 | F | c.1735G > A (p.Gly579Arg) | Missense | 10 | 4 | 9.9 | 3 | 568 |
| Px 81 | M | c.1404 + 2_1404+6del | Splice variant | 18 | 4 | 9.5 | 2.9 | 464 |
| Px 82 | M | c.208_212del (p.Val70Serfs*7) | Frameshift | 18 | 4 | 9.74 | 2.37 | 525 |
| Px 84 | F | c.416A > G (p.Tyr139Cys) | Missense | 8 | 2 | 9.9 | 3.7 | 728 |
| Px 91 | F | c.254G > C (p.Cys85Ser) | Missense | 11 | 4.5 | 10 | 2.7 | 442 |
| Px 92 | F | Deletion exon 14 | Frameshift | 7 | 5 | 9.1 | 2.8 | 578 |
| Px 93 | F | c.316del (p.Trp106Glyfs*2) | Frameshift | 9 | 5 | 9.7 | 2.6 | 417 |
| Px 94 | F | c.1699C > T (p.Arg567*) | Nonsense | 5 | 3.5 | 10.1 | 2.5 | 587 |
| Px 95 | F | c.1586_1586 + 1del (Splice site) | Splice variant | 12 | 5 | 10.1 | 3.2 | 854 |
| Px 96 | M | c.1735G > A (p.Gly579Arg) | Missense | 12 | 1.75 | 9 | 2.7 | 367 |
| Px 97 | F | c.1586+1G > A (Splice donor) | Splice variant | 13 | 3.5 | 9.9 | 2.4 | 389 |
| Px 98 | M | Deletion (Exon 12) | Frameshift | 14 | 1.75 | 9.7 | 2.1 | 1042 |
| Px 101 | F | c.1735G > A (p.Gly579Arg) | Missense | 14 | 2.5 | 9.5 | 2.9 | 527 |
| Px 102 | F | c.1173+1G > T (Splice donor) | Splice variant | 11 | 2.75 | 9.7 | 2.4 | 519 |
| Px 103 | M | c.933+1G > T (Splice donor) | Splice variant | 14 | 5 | 9.5 | 2.2 | 530 |
| Px 104 | F | c.1586+5G > A (Intronic) | Splice variant | 10 | 3 | 9.5 | 3.5 | 453 |
| Px 105 | M | c.2214_2234del (p.Met739_Ser745del) | Frameshift | 12 | 3.75 | 9.6 | 3.4 | 599 |
| Px 106 | M | c.2078G > A (p.Cys693Tyr) | Missense | 18 | 4 | 10.1 | 2.3 | 325 |
| Px 107 | F | c.1864T > C (p.Tyr622His) | Missense | 4 | 4.5 | 9.4 | 3 | 454 |
| Px 109 | F | c.1700G > C (p.Arg567Pro) | Missense | 7 | 5.5 | 9.8 | 2.7 | 507 |
| Px 112 | F | Deletion (Exons 10-11) | Frameshift | 5 | 3.75 | 9.2 | 2.7 | 485 |
| Px 116 | F | c.1444_1482+234del | Frameshift | 12 | 4.5 | 9.3 | 2.8 | 777 |
| Px 117 | F | c.1444_1482+234del | Frameshift | 13 | 4.5 | 9.5 | 3.2 | 726 |
| Px 128 | F | c.591A > G (Silent) | Silent | 20 | 1 | 9.6 | 2.2 | 293 |
| Px 129 | F | c.732+4_732 + 5insCA | Splice variant | 14 | 1.75 | 9.3 | 2.2 | 205 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Valdez, M.A.; Blanco-López, A.; Velázquez-Arestegui, D.; Vera-Zazueta, T.; Colmenares-Bonilla, D.; Reyes-Morales, L.; Blanco-Uriarte, M.A.; Monterde-Cruz, L.; Hidalgo-Bravo, A. Clinical, Radiographic, and Molecular Analysis of Patients with X-Linked Hypophosphatemic Rickets: Looking for Phenotype–Genotype Correlation. Diagnostics 2025, 15, 91. https://doi.org/10.3390/diagnostics15010091
Olivas-Valdez MA, Blanco-López A, Velázquez-Arestegui D, Vera-Zazueta T, Colmenares-Bonilla D, Reyes-Morales L, Blanco-Uriarte MA, Monterde-Cruz L, Hidalgo-Bravo A. Clinical, Radiographic, and Molecular Analysis of Patients with X-Linked Hypophosphatemic Rickets: Looking for Phenotype–Genotype Correlation. Diagnostics. 2025; 15(1):91. https://doi.org/10.3390/diagnostics15010091
Chicago/Turabian StyleOlivas-Valdez, Marco A., Armando Blanco-López, Daniela Velázquez-Arestegui, Teresita Vera-Zazueta, Douglas Colmenares-Bonilla, Lilian Reyes-Morales, Miguel A. Blanco-Uriarte, Lucero Monterde-Cruz, and Alberto Hidalgo-Bravo. 2025. "Clinical, Radiographic, and Molecular Analysis of Patients with X-Linked Hypophosphatemic Rickets: Looking for Phenotype–Genotype Correlation" Diagnostics 15, no. 1: 91. https://doi.org/10.3390/diagnostics15010091
APA StyleOlivas-Valdez, M. A., Blanco-López, A., Velázquez-Arestegui, D., Vera-Zazueta, T., Colmenares-Bonilla, D., Reyes-Morales, L., Blanco-Uriarte, M. A., Monterde-Cruz, L., & Hidalgo-Bravo, A. (2025). Clinical, Radiographic, and Molecular Analysis of Patients with X-Linked Hypophosphatemic Rickets: Looking for Phenotype–Genotype Correlation. Diagnostics, 15(1), 91. https://doi.org/10.3390/diagnostics15010091