
Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers
 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Plasma DNA Extraction
2.2. Amplification of HSATII Repeat Sequences from Plasma DNA
2.3. Sequencing of HSATII Amplicons
2.4. Bioinformatic Analysis
2.5. Statistical Analysis and Data Visualization
3. Results
3.1. High Fragmentation of cfDNA in Breast Cancer Patients Affects the Amplification of Longer HSATII Amplicons
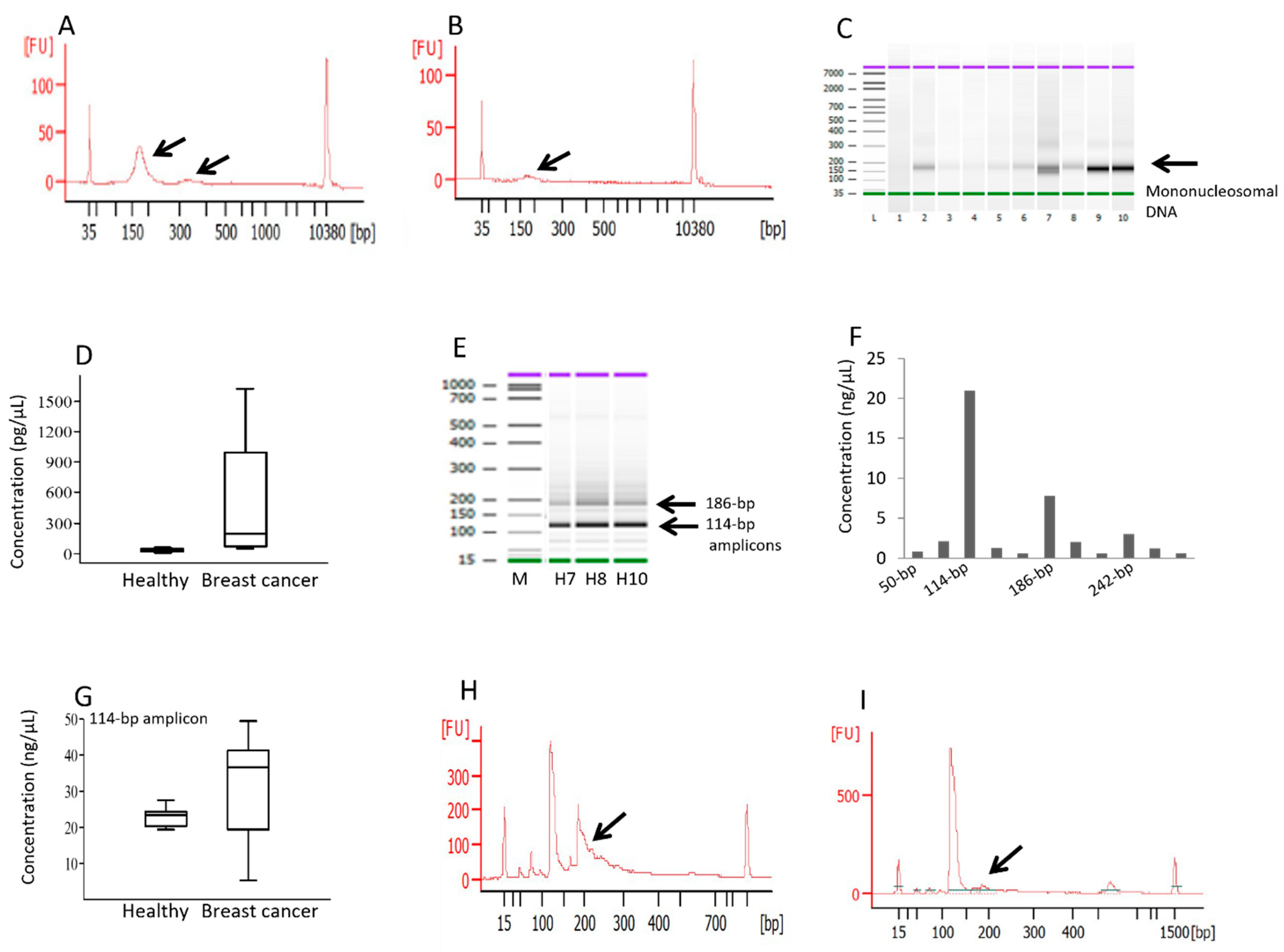
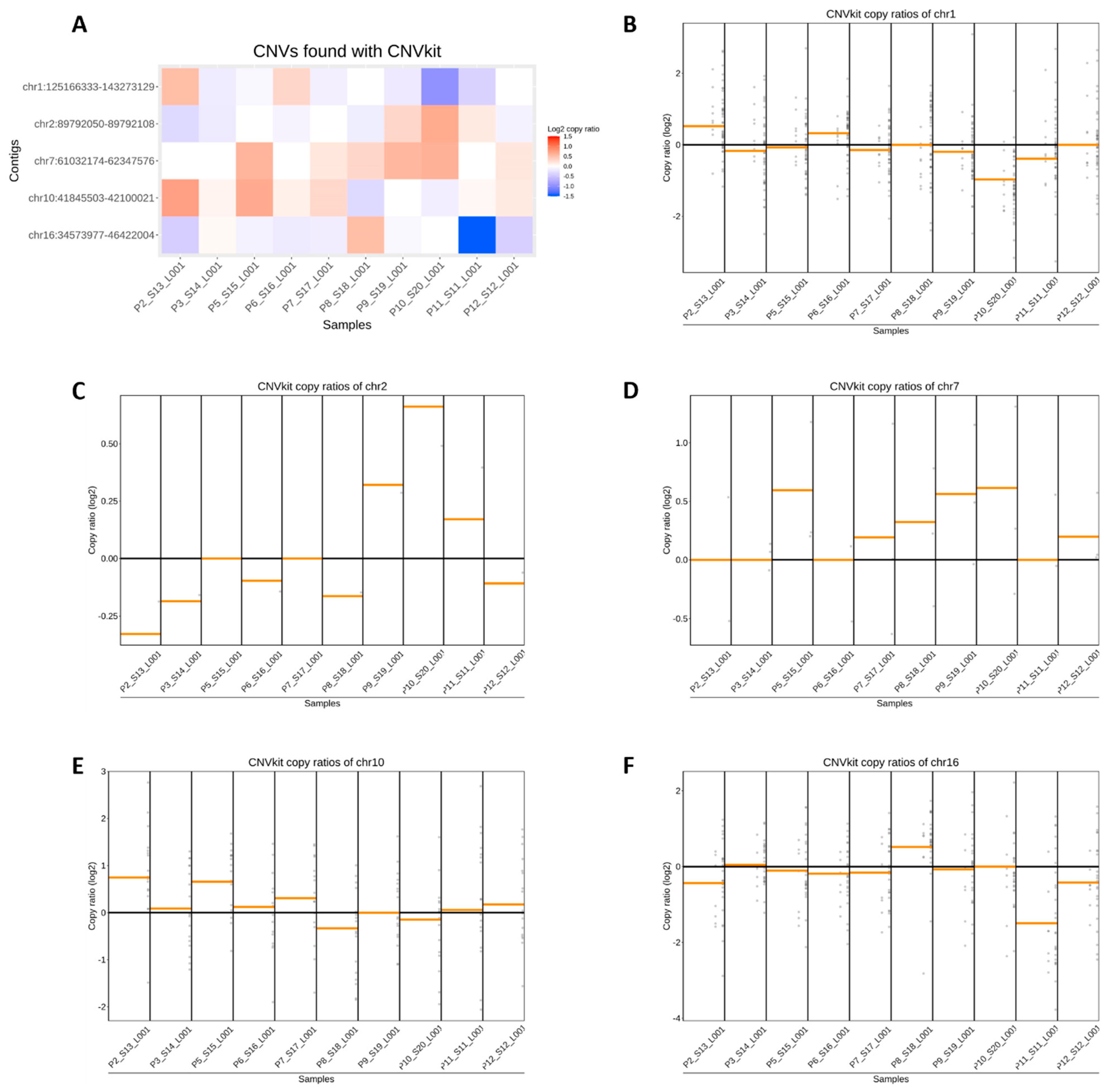
3.2. Estimation of Copy Ratios of HSATII in Breast Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2018, 20, 71–88. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Holdenrieder, S. The changing face of circulating tumor DNA (ctDNA) profiling: Factors that shape the landscape of methodologies, technologies, and commercialization. Med. Genet. 2023, 35, 201–235. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Peng, J.; Xiao, Q.; Wu, H.-X.; Wu, X.; Wang, F.; Li, L.; Ding, P.; Zhao, Q.; Li, Y. Postoperative circulating tumor DNA as markers of recurrence risk in stages II to III colorectal cancer. J. Hematol. Oncol. 2021, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Cuk, K.; Heil, J.; Golatta, M.; Schott, S.; Sohn, C.; Schneeweiss, A.; Burwinkel, B.; Surowy, H. Cell-free circulating DNA integrity is an independent predictor of impending breast cancer recurrence. Oncotarget 2017, 8, 54537–54547. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Offin, M.; Stephens, D.; Ni, A.; Lee, A.; Pavlakis, N.; Clarke, S.; Diakos, C.I.; Datta, S.; Tandon, N. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. JNCI J. Natl. Cancer Inst. 2019, 111, 575–583. [Google Scholar] [CrossRef]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.-S.; Mortensen, F.V.; Stribolt, K. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Davis, A.A.; Gerratana, L.; Velimirovic, M.; Shah, A.N.; Wehbe, F.; Katam, N.; Zhang, Q.; Flaum, L.; Siziopikou, K.P. The use of serial circulating tumor DNA to detect resistance alterations in progressive metastatic breast cancer. Clin. Cancer Res. 2021, 27, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Rohanizadegan, M. Analysis of circulating tumor DNA in breast cancer as a diagnostic and prognostic biomarker. Cancer Genet. 2018, 228–229, 159–168. [Google Scholar] [CrossRef]
- Ungerer, V.; Bronkhorst, A.J.; Uhlig, C.; Holdenrieder, S. Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line. Diagnostics 2022, 12, 1896. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Oberhofer, A.; Gabriel, S.; Polatoglou, E.; Randeu, H.; Uhlig, C.; Pfister, H.; Mayer, Z.; Holdenrieder, S. New Perspectives on the Importance of Cell-Free DNA Biology. Diagnostics 2022, 12, 2147. [Google Scholar] [CrossRef]
- Oberhofer, A.; Bronkhorst, A.J.; Uhlig, C.; Ungerer, V.; Holdenrieder, S. Tracing the Origin of Cell-Free DNA Molecules through Tissue-Specific Epigenetic Signatures. Diagnostics 2022, 12, 1834. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Bronkhorst, A.J.; Holdenrieder, S. The Utility of Repetitive Cell-Free DNA in Cancer Liquid Biopsies. Diagnostics 2022, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, Y.; Mouliere, F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019, 36, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.C.; Lo, Y.M.D. Cell-Free DNA Fragmentomics in Liquid Biopsy. Diagnostics 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Grabuschnig, S.; Soh, J.; Heidinger, P.; Bachler, T.; Hirschböck, E.; Rodriguez, I.R.; Schwendenwein, D.; Sensen, C.W. Circulating cell-free DNA is predominantly composed of retrotransposable elements and non-telomeric satellite DNA. J. Biotechnol. 2020, 313, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumour Biol. 2018, 40, 1010428318801190. [Google Scholar] [CrossRef]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Sunami, E.; Vu, A.T.; Nguyen, S.L.; Giuliano, A.E.; Hoon, D.S.B. Quantification of LINE1 in circulating DNA as a molecular biomarker of breast cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wu, W.; Han, L.; Yu, W.; Du, Y. A quantitative analysis of the potential biomarkers of non-small cell lung cancer by circulating cell-free DNA. Oncol. Lett. 2018, 16, 4353–4360. [Google Scholar] [CrossRef] [PubMed]
- Umetani, N.; Giuliano, A.E.; Hiramatsu, S.H.; Amersi, F.; Nakagawa, T.; Martino, S.; Hoon, D.S.B. Prediction of breast tumor progression by integrity of free circulating DNA in serum. J. Clin. Oncol. 2006, 24, 4270–4276. [Google Scholar] [CrossRef]
- Umetani, N.; Kim, J.; Hiramatsu, S.; Reber, H.A.; Hines, O.J.; Bilchik, A.J.; Hoon, D.S.B. Increased integrity of free circulating DNA in sera of patients with colorectal or periampullary cancer: Direct quantitative PCR for ALU repeats. Clin. Chem. 2006, 52, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Gang, F.; Li, X.; Jin, T.; Houbao, H.; Yu, C.; Guorong, L. Plasma cell-free DNA and its DNA integrity as biomarker to distinguish prostate cancer from benign prostatic hyperplasia in patients with increased serum prostate-specific antigen. Int. Urol. Nephrol. 2013, 45, 1023–1028. [Google Scholar] [CrossRef]
- Douville, C.; Springer, S.; Kinde, I.; Cohen, J.D.; Hruban, R.H.; Lennon, A.M.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Detection of aneuploidy in patients with cancer through amplification of long interspersed nucleotide elements (LINEs). Proc. Natl. Acad. Sci. USA 2018, 115, 1871–1876. [Google Scholar] [CrossRef]
- Douville, C.; Cohen, J.D.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Schoen, R.E.; Tie, J.; Gibbs, P. Assessing aneuploidy with repetitive element sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 4858–4863. [Google Scholar] [CrossRef]
- Nawroz, H.; Koch, W.; Anker, P.; Stroun, M.; Sidransky, D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat. Med. 1996, 2, 1035–1037. [Google Scholar] [CrossRef]
- Nagai, Y.; Sunami, E.; Yamamoto, Y.; Hata, K.; Okada, S.; Murono, K.; Yasuda, K.; Otani, K.; Nishikawa, T.; Tanaka, T. LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 2017, 8, 11906. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Pezer, Ž.; Brajković, J.; Feliciello, I.; Ugarković, Đ. Satellite DNA-mediated effects on genome regulation. Repetitive DNA 2012, 7, 153–169. [Google Scholar]
- Bersani, F.; Lee, E.; Kharchenko, P.V.; Xu, A.W.; Liu, M.; Xega, K.; MacKenzie, O.C.; Brannigan, B.W.; Wittner, B.S.; Jung, H. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15148–15153. [Google Scholar] [CrossRef] [PubMed]
- Özgür, E.; Mayer, Z.; Keskin, M.; Yörüker, E.E.; Holdenrieder, S.; Gezer, U. Satellite 2 repeat DNA in blood plasma as a candidate biomarker for the detection of cancer. Clin. Chim. Acta 2021, 514, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.O.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.W. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 October 2023).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H.; Chang, W.; Wickham, M.H. Package ‘ggplot2’. Creat. Elegant Data Vis. Using Gramm. Graph. Version 2016, 2, 1–189. [Google Scholar]
- Suwinski, P.; Ong, C.; Ling, M.H.; Poh, Y.M.; Khan, A.M.; Ong, H.S. Advancing personalized medicine through the application of whole exome sequencing and big data analytics. Front. Genet. 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Roch, B.; Mazard, T.; Blache, P.; Dache, Z.A.A.; Pastor, B.; Pisareva, E.; Tanos, R.; Thierry, A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021, 6, e144561. [Google Scholar] [CrossRef]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, S.; Raphael, B.J. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat. Commun. 2020, 11, 4301. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Hedtke, M.; Pessoa Rejas, R.; Froelich, M.F.; Ast, V.; Duda, A.; Mirbach, L.; Costina, V.; Martens, U.M.; Hofheinz, R.D.; Neumaier, M. Liquid profiling of circulating tumor DNA in colorectal cancer: Steps needed to achieve its full clinical value as standard care. Mol. Oncol. 2022, 16, 2042–2056. [Google Scholar] [CrossRef]
- Elshimali, Y.I.; Khaddour, H.; Sarkissyan, M.; Wu, Y.; Vadgama, J.V. The clinical Utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int. J. Mol. Sci. 2013, 14, 18925–18958. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Chromosomal Coordinates | Log2 Copy Ratio | Weight | Log2 Copy Ratio vs. Log2 Read Depth (Pearson’s r) | |
|---|---|---|---|---|
| Significant copy number gains | Chr1:143234645-143234759 | 1.98 | 0.29 | 0.65 |
| Chr16:46395162-46395250 | 1.32 | 0.57 | 0.21 | |
| Chr16:46398850-46398995 | 1.22 | 0.42 | 0.71 | |
| Chr10:42087568-42087682 | 1.19 | 0.43 | 0.47 | |
| Chr10:41850696-41850784 | 1.08 | 0.66 | 0.74 | |
| Chr1:125173758-125173872 | 1.02 | 0.85 | 0.40 | |
| Chr10:41845503-41845591 | 0.88 | 0.35 | 0.74 | |
| Chr1:143265528-143265702 | 0.78 | 0.49 | −0.19 | |
| Chr10:42093679-42093822 | 0.73 | 0.34 | 0.77 | |
| Chr1:143249938-143250076 | 0.65 | 0.74 | 0.60 | |
| Significant copy number losses | Chr16:34594210-34594279 | −0.96 | 0.31 | 0.85 |
| Chr10:42097372-42097590 | −0.96 | 0.93 | 0.87 | |
| Chr16:34591868-34591971 | −1.03 | 0.35 | 0.91 | |
| Chr16:46394749-46394886 | −1.16 | 0.39 | 0.79 | |
| Chr1:143256460-143256641 | −1.24 | 0.56 | −0.06 | |
| Chr1:143206616-143206759 | −1.29 | 0.63 | −0.68 | |
| Chr16:46390193-46390340 | −1.38 | 0.77 | 0.82 | |
| Chr16:46388855-46388979 | −1.48 | 0.17 | 0.83 | |
| Chr1:143235834-143235977 | −1.50 | 0.23 | 0.46 | |
| Chr16:34591728-34591842 | −1.58 | 0.87 | −0.19 |
| Chromosomal Coordinates | Total Gains | Total Losses | Log2 Copy Ratio | Weight | Copy Ratio vs. Read Depth Correlation (Pearson’s r) | |||
|---|---|---|---|---|---|---|---|---|
| Avg. | Std. Dev. | Avg. | Std. Dev. | |||||
| Significant copy number gains | Chr16:46395162-46395250 | 10 | 0 | 1.32 | 0.51 | 0.57 | 0.03 | 0.21 |
| Chr16:46398850-46398995 | 10 | 0 | 1.22 | 0.37 | 0.42 | 0.02 | 0.71 | |
| Chr16:46421913-46422004 | 10 | 0 | 0.49 | 0.33 | 0.76 | 0.03 | 0.61 | |
| Chr1:143234645-143234759 | 9 | 1 | 1.98 | 1.03 | 0.29 | 0.02 | 0.65 | |
| Chr10:42087568-42087682 | 9 | 1 | 1.19 | 0.84 | 0.43 | 0.02 | 0.47 | |
| Chr10:41850696-41850784 | 9 | 1 | 1.08 | 1.01 | 0.66 | 0.03 | 0.74 | |
| Chr10:41845503-41845591 | 9 | 1 | 0.88 | 1.17 | 0.35 | 0.03 | 0.74 | |
| Chr1:143265528-143265702 | 9 | 1 | 0.78 | 0.63 | 0.49 | 0.02 | −0.19 | |
| Chr1:143244455-143244541 | 9 | 1 | 0.64 | 0.47 | 0.27 | 0.03 | 0.49 | |
| Chr16:34588502-34588580 | 9 | 1 | 0.58 | 0.38 | 0.70 | 0.03 | 0.77 | |
| Chr16:46389286-46389443 | 9 | 1 | 0.56 | 0.38 | 0.43 | 0.02 | 0.71 | |
| Significant copy number losses | Chr16:46394749-46394886 | 1 | 9 | −0.63 | 0.77 | 0.66 | 0.02 | 0.64 |
| Chr1:125166914-125167051 | 1 | 9 | −0.64 | 0.53 | 0.20 | 0.02 | 0.89 | |
| Chr16:34573977-34574077 | 1 | 9 | −0.68 | 0.89 | 0.32 | 0.02 | 0.73 | |
| Chr1:143239360-143239536 | 1 | 9 | −0.74 | 0.86 | 0.46 | 0.02 | 0.20 | |
| Chr16:34591868-34591971 | 1 | 9 | −0.78 | 0.77 | 0.52 | 0.02 | 0.87 | |
| Chr16:46390193-46390340 | 1 | 9 | −1.29 | 0.85 | 0.63 | 0.02 | −0.68 | |
| Chr16:34591728-34591842 | 1 | 9 | −1.58 | 1.02 | 0.87 | 0.02 | −0.19 | |
| Chr16:46390975-46391112 | 0 | 10 | −0.96 | 0.47 | 0.31 | 0.02 | 0.85 | |
| Chr1:143206616-143206759 | 0 | 10 | −1.24 | 0.77 | 0.56 | 0.02 | −0.06 | |
| Chr1:143235834-143235977 | 0 | 10 | −1.38 | 0.55 | 0.77 | 0.02 | 0.82 | |
| Chr10:42099756-42099907 | 0 | 10 | −1.48 | 0.38 | 0.17 | 0.02 | 0.83 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gezer, U.; Oberhofer, A.; Worf, K.; Stoetzer, O.; Holdenrieder, S.; Bronkhorst, A. Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers. Diagnostics 2024, 14, 609. https://doi.org/10.3390/diagnostics14060609
Gezer U, Oberhofer A, Worf K, Stoetzer O, Holdenrieder S, Bronkhorst A. Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers. Diagnostics. 2024; 14(6):609. https://doi.org/10.3390/diagnostics14060609
Chicago/Turabian StyleGezer, Ugur, Angela Oberhofer, Karolina Worf, Oliver Stoetzer, Stefan Holdenrieder, and Abel Bronkhorst. 2024. "Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers" Diagnostics 14, no. 6: 609. https://doi.org/10.3390/diagnostics14060609
APA StyleGezer, U., Oberhofer, A., Worf, K., Stoetzer, O., Holdenrieder, S., & Bronkhorst, A. (2024). Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers. Diagnostics, 14(6), 609. https://doi.org/10.3390/diagnostics14060609