
Clinical Efficacy and Safety of an Automatic Closed-Suction System in Mechanically Ventilated Patients with Pneumonia: A Multicenter, Prospective, Randomized, Non-Inferiority, Investigator-Initiated Trial

 , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Intervention and Randomization
2.3. Outcomes
2.4. Sample Size Calculation
2.5. Statistical Analysis
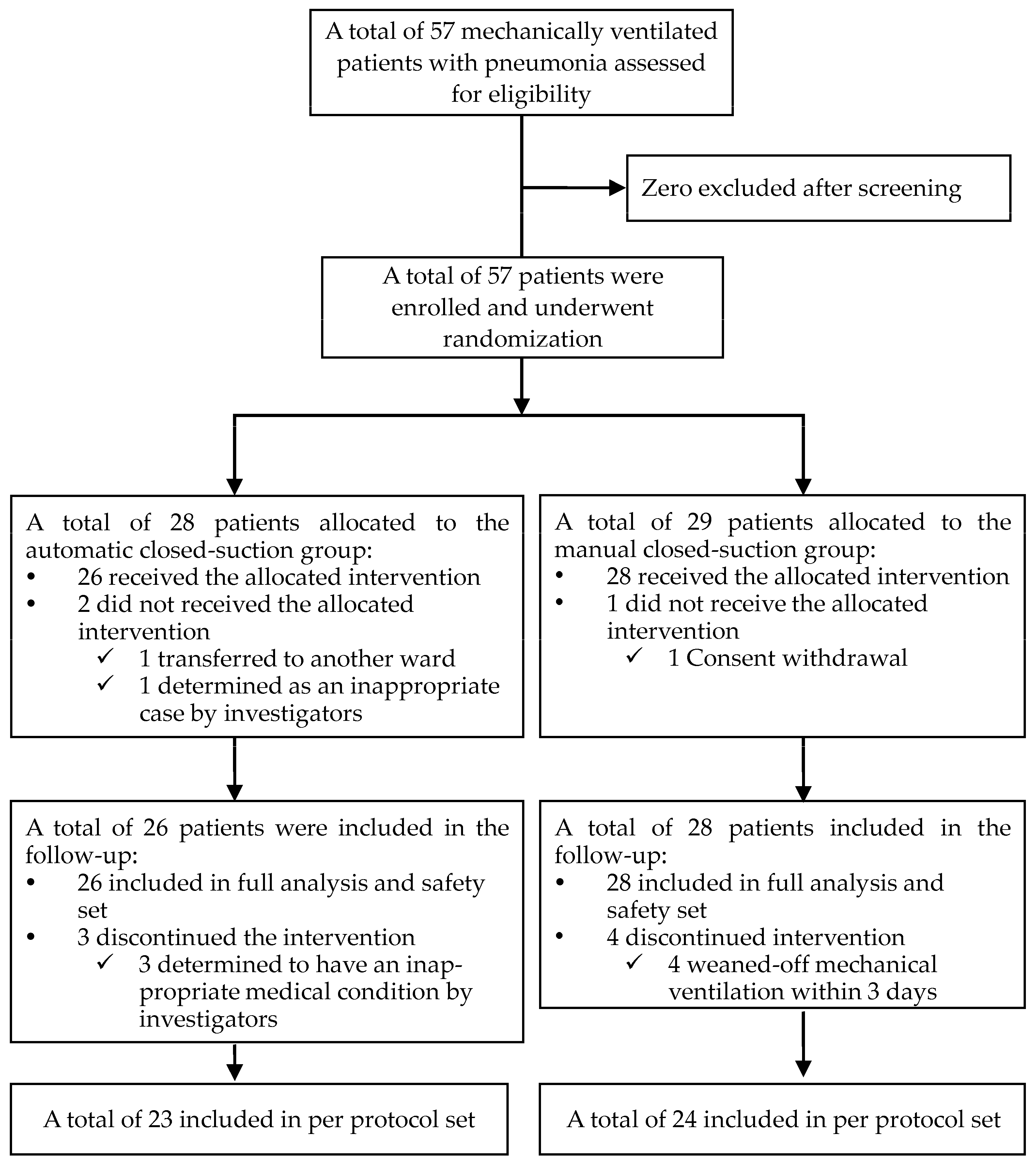
3. Results
3.1. Participant Characteristics
3.2. Efficacy Outcomes
3.3. Safety Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Blakeman, T.C.; Scott, J.B.; Yoder, M.A.; Capellari, E.; Strickland, S.L. AARC Clinical Practice Guidelines: Artificial Airway Suctioning. Respir. Care 2022, 67, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Mwakanyanga, E.T.; Masika, G.M.; Tarimo, E.A.M. Intensive care nurses’ knowledge and practice on endotracheal suctioning of the intubated patient: A quantitative cross-sectional observational study. PLoS ONE 2018, 13, e0201743. [Google Scholar] [CrossRef] [PubMed]
- American Association for Respiratory Care. AARC Clinical Practice Guidelines. Endotracheal suctioning of mechanically ventilated patients with artificial airways 2010. Respir. Care 2010, 55, 758–764. [Google Scholar]
- Pedersen, C.M.; Rosendahl-Nielsen, M.; Hjermind, J.; Egerod, I. Endotracheal suctioning of the adult intubated patient--what is the evidence? Intensive Crit. Care Nurs. 2009, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hu, S.; Liu, X.; Wang, N.; Zhao, J.; Liu, P.; Chen, K.; Hu, J. Intensive care nurses’ knowledge and practice of evidence-based recommendations for endotracheal suctioning: A multisite cross-sectional study in Changsha, China. BMC Nurs. 2021, 20, 186. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.K. The nursing shortage. Past, present, and future. J. Nurs. Adm. 2002, 32, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Lopez, V.; Anderson, J.; West, S.; Cleary, M. Does the COVID-19 Pandemic Further Impact Nursing Shortages? Issues Ment. Health Nurs. 2022, 43, 293–295. [Google Scholar] [CrossRef]
- Suh, D.W.; Lee, S.B.; Kim, S.M. Study on Device System to Reduce Tracheal Mucosal Injury in Intubation Patients. J. Med. Devices 2022, 16, 031006. [Google Scholar] [CrossRef]
- Jeong, J.H.; Nam, S.J.; Cho, Y.J.; Lee, Y.J.; Kim, S.J.; Song, I.A.; Park, S.H.; Jeon, Y.T. A Closed-Suction Catheter with a Pressure Valve Can Reduce Tracheal Mucosal Injury in Intubated Patients. Acute Crit. Care 2014, 29, 7–12. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kim, H.S.; Yang, H.J.; Lee, Y.J.; Park, J.S.; Yoon, H.I.; Kim, H.B.; Yim, J.J.; Lee, J.H.; Lee, C.T.; et al. Pilot Study of Aerosolised Plus Intravenous Vancomycin in Mechanically Ventilated Patients with Methicillin-Resistant Staphylococcus aureus Pneumonia. J. Clin. Med. 2020, 9, 476. [Google Scholar] [CrossRef]
- Lee, H.W.; Min, J.; Park, J.; Lee, Y.J.; Kim, S.J.; Park, J.S.; Yoon, H.I.; Lee, J.H.; Lee, C.T.; Cho, Y.J. Clinical impact of early bronchoscopy in mechanically ventilated patients with aspiration pneumonia. Respirology 2015, 20, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.L.; Kearney, P.A.; Johnson, S.B.; Niblett, J.B.; MacMillan, N.L.; McClain, R.E. Closed versus open endotracheal suctioning: Costs and physiologic consequences. Crit. Care Med. 1994, 22, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Lasocki, S.; Lu, Q.; Sartorius, A.; Fouillat, D.; Remerand, F.; Rouby, J.J. Open and closed-circuit endotracheal suctioning in acute lung injury: Efficiency and effects on gas exchange. Anesthesiology 2006, 104, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, B.; Webb, C.H. Closed tracheal suctioning systems and infection control in the intensive care unit. J. Hosp. Infect. 1998, 39, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Ugras, G.A.; Aksoy, G. The effects of open and closed endotracheal suctioning on intracranial pressure and cerebral perfusion pressure: A crossover, single-blind clinical trial. J. Neurosci. Nurs. 2012, 44, E1–E8. [Google Scholar] [CrossRef] [PubMed]
- Jongerden, I.P.; Buiting, A.G.; Leverstein-van Hall, M.A.; Speelberg, B.; Zeidler, S.; Kesecioglu, J.; Bonten, M.J. Effect of open and closed endotracheal suctioning on cross-transmission with Gram-negative bacteria: A prospective crossover study. Crit. Care Med. 2011, 39, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- David, D.; Samuel, P.; David, T.; Keshava, S.N.; Irodi, A.; Peter, J.V. An open-labelled randomized controlled trial comparing costs and clinical outcomes of open endotracheal suctioning with closed endotracheal suctioning in mechanically ventilated medical intensive care patients. J. Crit. Care 2011, 26, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Akerman, E.; Larsson, C.; Ersson, A. Clinical experience and incidence of ventilator-associated pneumonia using closed versus open suction-system. Nurs. Crit. Care 2014, 19, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hamishekar, H.; Shadvar, K.; Taghizadeh, M.; Golzari, S.E.; Mojtahedzadeh, M.; Soleimanpour, H.; Mahmoodpoor, A. Ventilator-associated pneumonia in patients admitted to intensive care units, using open or closed endotracheal suctioning. Anesth. Pain Med. 2014, 4, e21649. [Google Scholar] [CrossRef]
- Deppe, S.A.; Kelly, J.W.; Thoi, L.L.; Chudy, J.H.; Longfield, R.N.; Ducey, J.P.; Truwit, C.L.; Antopol, M.R. Incidence of colonization, nosocomial pneumonia, and mortality in critically ill patients using a Trach Care closed-suction system versus an open-suction system: Prospective, randomized study. Crit. Care Med. 1990, 18, 1389–1393. [Google Scholar] [CrossRef]
- Kalil, A.C.; Metersky, M.L.; Klompas, M.; Muscedere, J.; Sweeney, D.A.; Palmer, L.B.; Napolitano, L.M.; O’Grady, N.P.; Bartlett, J.G.; Carratala, J.; et al. Management of Adults with Hospital-acquired and Ventilator-associated Pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin. Infect. Dis. 2016, 63, e61–e111. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Variables | Automatic Closed Suction | Manual Closed Suction | Total | |
|---|---|---|---|---|
| (n = 26) | (n = 28) | (n = 54) | ||
| Age, years | 66.1 ± 18.1 | 71.5 ± 10.9 | 68.9 ± 14.9 | |
| Sex, male | 22 (84.6%) | 22 (78.6%) | 44 (81.5%) | |
| Height, cm | 164.0 ± 7.6 | 165.2 ± 9.4 | 164.7 ± 8.5 | |
| Weight, kg | 61.4 ± 12.6 | 59.4 ± 15.6 | 60.4 ± 14.2 | |
| Duration of pneumonia, days | 4.0 (1.0–9.0) | 5.0 (2.5–11.0) | 4.0 (1.0–9.0) | |
| Duration in ICU, days | 2.0 (2.0–5.0) | 2.0 (1.0–6.0) | 2.0 (1.0–6.0) | |
| APACHE II score | 19.7 ± 5.6 | 20.9 ± 4.1 | 20.3 ± 4.8 | |
| Chest radiography | ||||
| Diffuse infiltration | 22 (84.6%) | 24 (85.7%) | 46 (85.2%) | |
| Localized infiltration | 4 (15.4%) | 4 (14.3%) | 8 (14.8%) | |
| ARDS | 15 (57.7%) | 15 (53.6%) | 30 (55.6%) | |
| Hemodynamic variables | ||||
| Mean blood pressure, mmHg | 87 ± 14 | 90 ± 16 | 89 ± 15 | |
| Heart rate, /min | 92 ± 22 | 97 ± 22 | 95 ± 22 | |
| Respiratory rate, /min | 21 ± 5 | 22 ± 4 | 22 ± 5 | |
| Body temperature, °C | 36.9 ± 0.6 | 36.8 ± 0.6 | 36.9 ± 0.6 | |
| SpO2, % | 97 ± 2 | 97 ± 3 | 97 ± 3 | |
| Variables | Automatic Closed Suction | Manual Closed Suction | p-Value |
|---|---|---|---|
| (n = 23) | (n = 24) | ||
| Primary efficacy outcome | |||
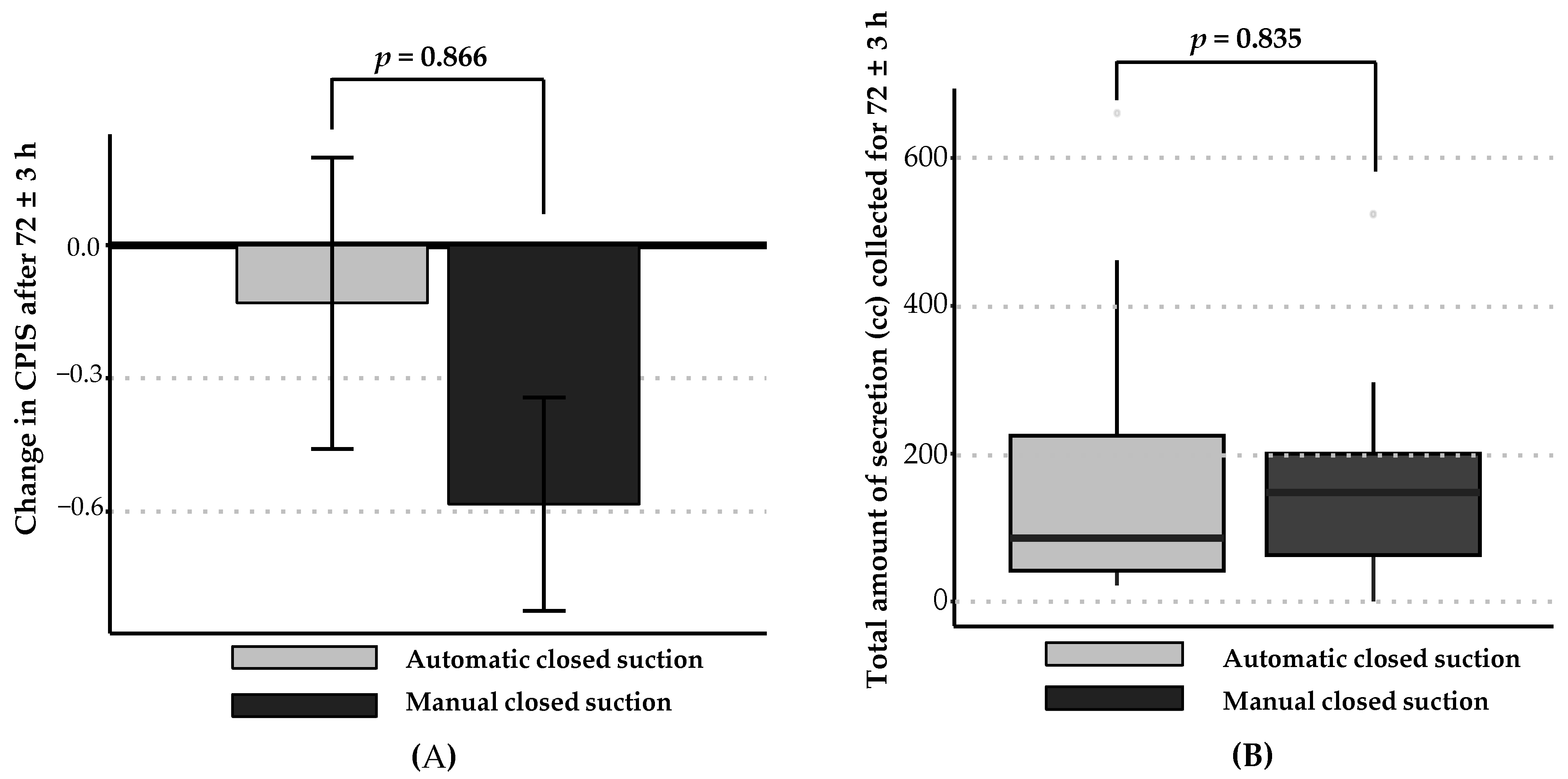
| Change in CPIS = (CPIS after 72 h − baseline CPIS) | 0.866 | ||
| Number | 23 | 24 | |
| Mean ± SD | −0.13 ± 1.58 | −0.58 ± 1.18 | |
| Mean difference (intervention—control) (97.5% confidence interval for difference) | 0.45 (−∞, 1.27) | ||
| Secondary efficacy outcomes | |||
| Modified CPIS at baseline | 3.39 ± 1.78 | 3.75 ± 1.92 | 0.531 |
| Modified CPIS at 72 ± 3 h | 3.26 ± 1.48 | 3.17 ± 1.58 | 0.760 |
| * Improvement rate in modified CPIS | 9 (39.13%) | 13 (54.17%) | 0.385 |
| Number of suctions performed in addition to those specified in the protocol | 7.0 (3.5–12.0) | 4.0 (2.0–7.5) | 0.189 |
| Total amount of secretions collected, cc | 86.0 (42.0–247.0) | 147.5 (48.5–207.5) | 0.835 |
| † Device satisfaction survey after 72 h | 5.88 ± 1.05 | ||
| Variables | Automatic Closed Suction | Manual Closed Suction | Total |
|---|---|---|---|
| (n = 26) | (n = 28) | (n = 54) | |
| Treatment-emergent adverse events | 3 (11.54%) | 0 (0.00%) | 3 (5.56%) |
| Mild (atrial fibrillation) | 1 (3.85%) | 0 (0.00%) | 1 (1.85%) |
| Moderate | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| Severe (cardiac arrest) | 2 (7.69%) | 0 (0.00%) | 2 (3.70%) |
| Device-related adverse reactions | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| Complication rate (%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| Tracheal mucosal injury incidence (%) | 1 (3.85%) | 1 (3.57%) | 2 (3.70%) |
| p = 1.000 | |||
| Tracheal mucosal injury improvement (%) | 12 (46.15%) | 6 (21.43%) | 18 (33.33%) |
| p = 0.102 | |||
| Device malfunction rate | 1 (3.85%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joo, D.-H.; Park, H.C.; Kim, J.H.; Yang, S.H.; Kim, T.H.; Kim, H.-J.; Song, M.J.; Lim, S.Y.; Kim, S.A.; Bae, H.W.; et al. Clinical Efficacy and Safety of an Automatic Closed-Suction System in Mechanically Ventilated Patients with Pneumonia: A Multicenter, Prospective, Randomized, Non-Inferiority, Investigator-Initiated Trial. Diagnostics 2024, 14, 1068. https://doi.org/10.3390/diagnostics14111068
Joo D-H, Park HC, Kim JH, Yang SH, Kim TH, Kim H-J, Song MJ, Lim SY, Kim SA, Bae HW, et al. Clinical Efficacy and Safety of an Automatic Closed-Suction System in Mechanically Ventilated Patients with Pneumonia: A Multicenter, Prospective, Randomized, Non-Inferiority, Investigator-Initiated Trial. Diagnostics. 2024; 14(11):1068. https://doi.org/10.3390/diagnostics14111068
Chicago/Turabian StyleJoo, Dong-Hyun, Hyo Chan Park, Joon Han Kim, Seo Hee Yang, Tae Hun Kim, Hyung-Jun Kim, Myung Jin Song, Sung Yoon Lim, Sung A Kim, Hee Won Bae, and et al. 2024. "Clinical Efficacy and Safety of an Automatic Closed-Suction System in Mechanically Ventilated Patients with Pneumonia: A Multicenter, Prospective, Randomized, Non-Inferiority, Investigator-Initiated Trial" Diagnostics 14, no. 11: 1068. https://doi.org/10.3390/diagnostics14111068
APA StyleJoo, D.-H., Park, H. C., Kim, J. H., Yang, S. H., Kim, T. H., Kim, H.-J., Song, M. J., Lim, S. Y., Kim, S. A., Bae, H. W., Ahn, Y. H., Yoon, S. M., Park, J., Lee, H. Y., Lee, J., Lee, S.-M., Lee, J. C., & Cho, Y.-J. (2024). Clinical Efficacy and Safety of an Automatic Closed-Suction System in Mechanically Ventilated Patients with Pneumonia: A Multicenter, Prospective, Randomized, Non-Inferiority, Investigator-Initiated Trial. Diagnostics, 14(11), 1068. https://doi.org/10.3390/diagnostics14111068