
DCDC2-Related Ciliopathy: Report of Six Polish Patients, Novel DCDC2 Variant, and Literature Review of Reported Cases

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Liver Biopsy
2.3. Molecular Analysis
2.4. Literature Search
3. Results
3.1. Patients’ Presentation
3.2. Literature Review
4. Discussion
5. Conclusions
- The main clinical presentation of DCDC2-related ciliopathy is liver disease in the form of neonatal sclerosing cholangitis.
- The predominance of early and severe liver disease associated with no or mildly expressed kidney involvement is observed in DCDC2-related ciliopathy.
- Our findings expand the molecular spectrum of pathogenic DCDC2 variants, provide a more accurate picture of the phenotypic expression associated with molecular changes in this gene and confirm a loss of functional behaviour as the mechanism of disease.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rock, N.; McLin, V. Liver involvement in children with ciliopathies. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Gunay-Aygun, M. Liver and kidney disease in ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151C, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, J.M.; Ahn, Y.H.; Kang, H.G.; Ha, I.I.; Lee, J.H.; Park, Y.S.; Kim, N.K.; Park, W.Y.; Cheong, H.I. Hepatorenal fibrocystic diseases in children. Pediatr. Nephrol. 2016, 31, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Wicher, D.; Obrycki, Ł.; Jankowska, I. Autosomal Recessive Polycystic Kidney Disease-The Clinical Aspects and Diagnostic Challenges. J. Pediatr. Genet. 2021, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stokman, M.; Lilien, M.; Knoers, N. Nephronophthisis-Related Ciliopathies 2016 [updated 2023 Mar 2]. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lipiński, P.; Ciara, E.; Jurkiewicz, D.; Pollak, A.; Wypchło, M.; Płoski, R.; Cielecka-Kuszyk, J.; Socha, P.; Pawłowska, J.; Jankowska, I. Targeted Next-Generation Sequencing in Diagnostic Approach to Monogenic Cholestatic Liver Disorders-Single-Center Experience. Front. Pediatr. 2020, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Bizet, A.A.; Lachaux, A.; Gonzales, E.; Filhol, E.; Collardeau-Frachon, S.; Jeanpierre, C.; Henry, C.; Fabre, M.; Viremouneix, L.; et al. DCDC2 Mutations Cause Neonatal Sclerosing Cholangitis. Hum. Mutat. 2016, 37, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Grammatikopoulos, T.; Sambrotta, M.; Strautnieks, S.; Foskett, P.; Knisely, A.S.; Wagner, B.; Deheragoda, M.; Starling, C.; Mieli-Vergani, G.; Smith, J.; et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J. Hepatol. 2016, 65, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Lu, Y.; Qiu, Y.L.; Wang, J.S. Neonatal sclerosing cholangitis caused by DCDC2 variations in two siblings and literature review. Zhonghua Er Ke Za Zhi 2018, 56, 623–627. [Google Scholar] [PubMed]
- Slater, B.; Bekheirnia, N.; Angelo, J.; Bi, W.; Braun, M.C.; Bekheirnia, M.R. Nephronophthisis due to a novel DCDC2 variant in a patient from African-Caribbean descent: A case report. Am. J. Med. Genet. A 2020, 182, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Vogel, G.F.; Maurer, E.; Entenmann, A.; Straub, S.; Knisely, A.S.; Janecke, A.R.; Müller, T. Co-existence of ABCB11 and DCDC2 disease: Infantile cholestasis requires both next-generation sequencing and clinical-histopathologic correlation. Eur. J. Hum. Genet. 2020, 28, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, J.; Li, X.; Zheng, D.; Yu, X.; Liu, Y.; Lan, F.; Wang, Z. Biallelic mutations in DCDC2 cause neonatal sclerosing cholangitis in a Chinese family. Clin. Res. Hepatol. Gastroenterol. 2020, 44, e103–e108. [Google Scholar] [CrossRef] [PubMed]
- Syryn, H.; Hoorens, A.; Grammatikopoulos, T.; Deheragoda, M.; Symoens, S.; Vande Velde, S.; Van Biervliet, S.; Van Winckel, M.; Verloo, P.; Callewaert, B.; et al. Two cases of DCDC2-related neonatal sclerosing cholangitis with developmental delay and literature review. Clin. Genet. 2021, 100, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Teker Düztaş, D.; Sarı, S.; Eğritaş Gürkan, Ö.; Kayhan, G.; Dalgıç, A.; Dalgıç, B. Two Cases With Neonatal Cholestasis and Renal Disorders Due to DCDC2 Mutation. Exp. Clin. Transplant. 2022, 20, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Fang, Y.; Wang, J.S.; Wang, Y.Z.; Zhang, Y.; Abuduxikuer, K.; Chen, L. Neonatal sclerosing cholangitis with novel mutations in DCDC2 (doublecortin domain-containing protein 2) in Chinese children. Front. Pediatr. 2023, 11, 1094895. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.omim.org/entry/605755 (accessed on 1 March 2023).
- Amedee-Manesme, O.; Bernard, O.; Brunelle, F.; Hadchouel, M.; Polonovski, C.; Baudon, J.J.; Beguet, P.; Alagille, D. Sclerosing cholangitis with neonatal onset. J. Pediatr. 1987, 111, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Hadj-Rabia, S.; Baala, L.; Vabres, P.; Hamel-Teillac, D.; Jacquemin, E.; Fabre, M.; Lyonnet, S.; De Prost, Y.; Munnich, A.; Hadchouel, M.; et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology 2004, 127, 1386–1390. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Hadj-Rabia, S.; Hamel-Teillac, D.; Hadchouel, M.; Prost, C.; Leal, S.M.; Jacquemin, E.; Sefiani, A.; De Prost, Y.; Courtois, G.; et al. Homozygosity mapping of a locus for a novel syndromic ichthyosis to chromosome 3q27-q28. J. Invest. Dermatol. 2002, 119, 70–76. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient/ Gender | Origin | Age at First Presentation | Presenting Signs and Symptoms | Intrahepatic Cholangiopathy on MRCP | Liver Phenotype, Including Histology | LTx/ Age at LTx | Follow-Up | Other | Genotype |
|---|---|---|---|---|---|---|---|---|---|
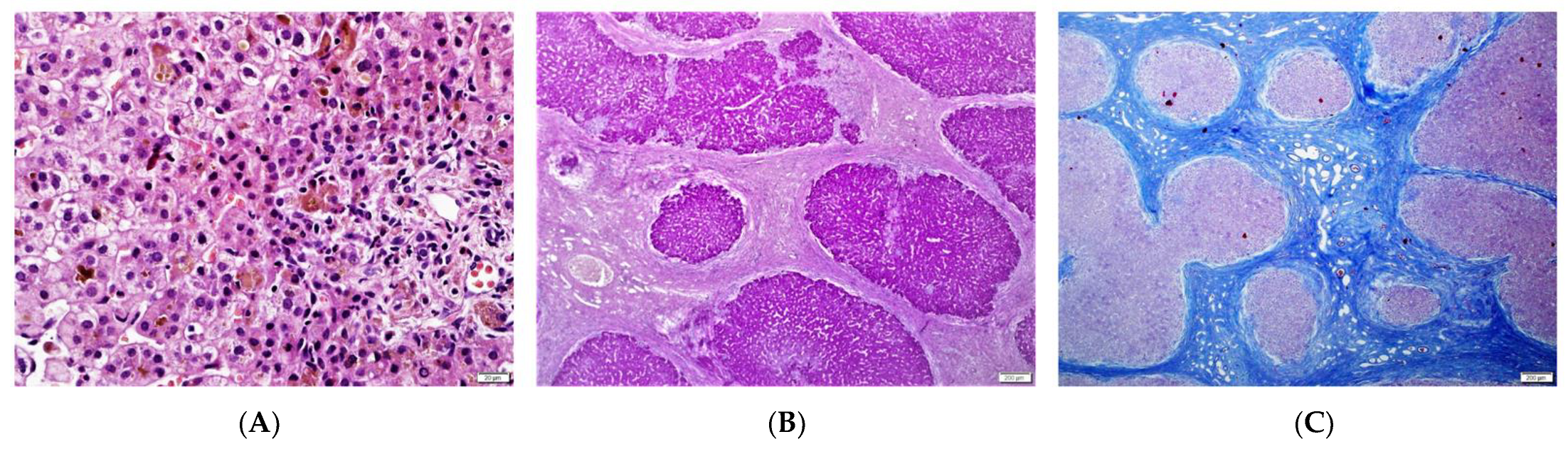
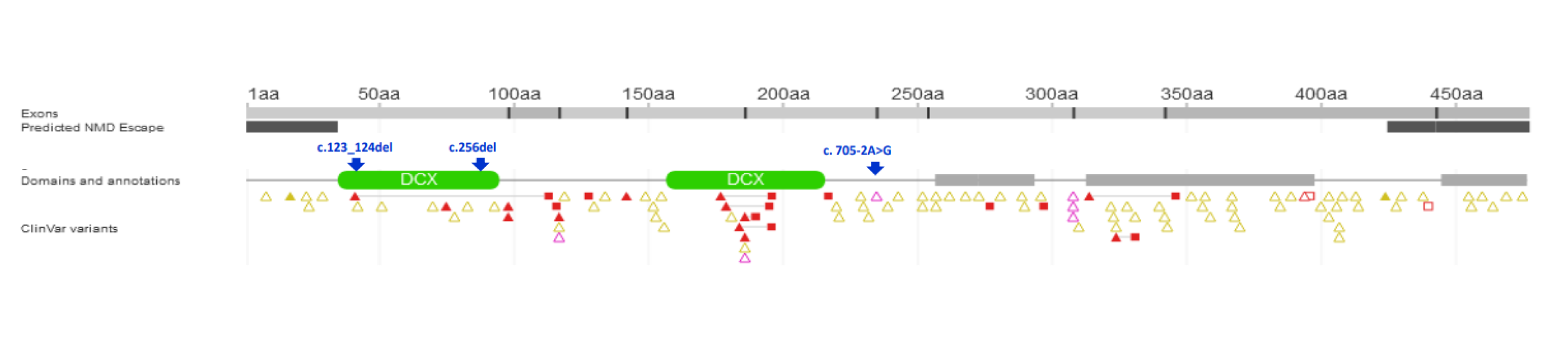
| 1/F | Caucasian | 25th day of life | Jaundice, acholic stools, elevated GGT | No | 2 m—liver biopsy: severe cholestasis, porto-portal fibrosis, moderate giant cell transformation of hepatocytes, no proliferation of bile ductules; 9 m—1st episode of oesophageal varices bleeding; 3.5 y—liver biopsy: mild cholestasis without biliary plugs, ductopenia, severe liver fibrosis; 5 y—2 episodes of oesophageal varices bleeding; 6 y—LTx with splenectomy, liver cirrhosis in the hepatic explant. | Yes/6 y | 8 y | Normal kidney function | c.[123_124del];[256del], p.(Ser42Glnfs*72)/ p.(Tyr86Thrfs*17) compound heterozygote |
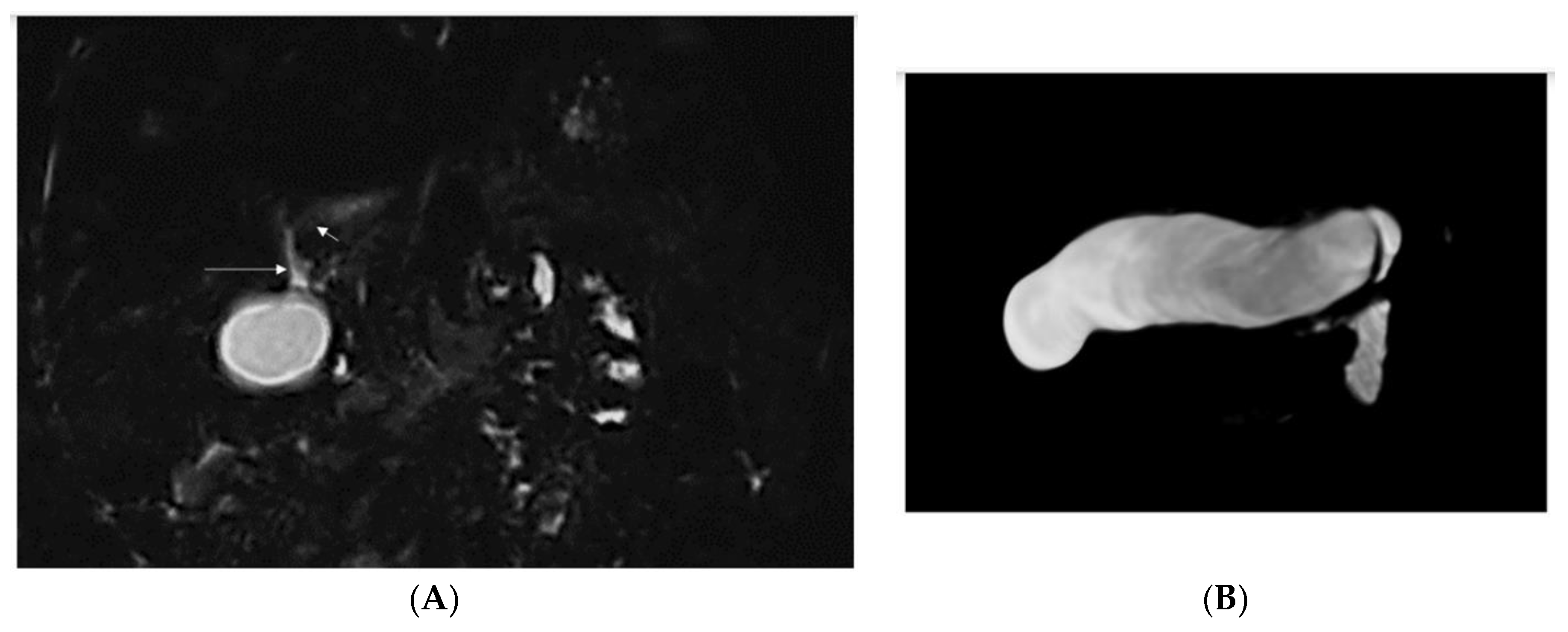
| 2/F Sister of Pt 1 | Caucasian | 2nd day of life | Jaundice, acholic stools, elevated GGT | Yes (8 mo) See Figure 1 | No liver biopsy | No | 9 m | right vesico-ureteral reflux II degree, persistent urachus | c.[123_124del];[256del], p.(Ser42Glnfs*72)/ p.(Tyr86Thrfs*17) compound heterozygote |
| 3/F | Caucasian | 2 m | Jaundice, elevated GGT | n.a. | 2 m—liver biopsy: mild cholestasis, portal fibrosis, focal ductular proliferation; 4 y—1st episode of oesophageal varices bleeding, ascites. | No | 12.5 y | Normal kidney function | c.[123_124del];[705-2A>G] p.(Ser42Glnfs*72)/ p.? compound heterozygote |
| 4/M Brother of Pt 3 and 5 | Caucasian | 2 m | Jaundice, elevated GGT | n.a. | 4 m—liver biopsy: moderate cholestasis, portal fibrosis, focal ductular proliferation; 12 m—oesophageal varices in gastroscopy. | No | 10 y | Normal kidney function | c.[123_124del];[705-2A>G] p.(Ser42Glnfs*72)/ p.? compound heterozygote |
| 5/M Brother of Pt 3 and 4 | Caucasian | 2 m | Jaundice, elevated GGT | n.a. | No liver biopsy | No | 5 y | Normal kidney function | c.[123_124del];[705-2A>G] p.(Ser42Glnfs*72)/ p.? compound heterozygote |
| 6/M | Caucasian | 3 m | Jaundice, elevated GGT | No | 5 m—liver biopsy—severe cholestasis with bile plugs, ductular proliferation; 2 y—oesophageal varices in gastroscopy; 2.5 y—liver biopsy: moderate cholestasis, severe fibrosis, ductopenia; 13 y—abdominal MR—liver cirrhosis, FNH. | No | 24 y | Normal kidney function | c.[123_124del];[ 123_124del] p.(Ser42Glnfs*72)/ p.(Ser42Glnfs*72) homozygote |
| Patient Age at Liver Biopsy | Fibrosis | Cholestasis | Ductular Changes | Inflammation | Giant Cell Transformation |
|---|---|---|---|---|---|
| Patient 1 | |||||
| 2 m | porto-portal fibrosis | ductal, acinar bile plugs, severe cholestasis | no | mild portal | moderate |
| 4.5 y | severe fibrosis | mild, hepatocellular | ductopenia | no | No |
| hepatic explant (6 y) | micronodular cirrhosis | mild cholestasis | diffuse ductular proliferation | mild portal | No |
| Patient 3 | |||||
| 2 m | portal fibrosis | mild, hepatocellular | focal ductular proliferation | mild portal, ductitis | No |
| Patient 4 | |||||
| 4 m | portal fibrosis | moderate hepatocellular | focal ductular proliferation | mild portal, ductitis | no |
| Patient 6 | |||||
| 5 m | periportal, bridging fibrosis | ductal, acinar bile plugs, severe cholestasis | ductular proliferation, focal DPM | mild portal | no |
| 2.5 y | severe fibrosis | ductular bile plugs, moderate hepatocellular | ductopenia, ductular proliferation | no | no |
| Patient/ Gender | Origin/ Consanguinity | Age at First Presentation | Presenting Signs and Symptoms | Intrahepatic Cholangiopathy on ERCP/MRCP | Liver Phenotype, Including Histology | LTx/ Age at LTx | Follow-Up | Other | Genetic Result for NM_016356.5(DCDC2) RefSeq | References |
|---|---|---|---|---|---|---|---|---|---|---|
| 1/n.a. | UK/Yes | n.a. | n.a. | n.a. | Hepatosplenomegaly, extensive fibrosis (11 mo) with destruction of bile ducts, bile focal duct proliferation with cholestasis | No | Died at 16 y from GI bleeding | Increased echogenicity, severe interstitial fibrosis, tubular dilation with prominent epithelial luminal budding, ESRD at 14 y | c.649A>T p.(Lys217*)/ c.649A>T p.(Lys217*) | Schueler et al., 2015 [8] |
| 2/n.a. | Czech/No | n.a. | n.a. | n.a. | Hepatosplenomegaly, ductal plate malformation, hepatic fibrosis, scant cholestasis | Yes/2 y | current age 9 y | No renal involvement | c.123_124del, p.(Ser42Glnfs*72)/ c.349-2A>G, p.(Val117Leufs*54) | |
| 3/F | Asian/Yes | 20 wk | Jaundice, acholic stools, GGT 247 IU/L | Yes | n.a. | No/ listed | Died at 16 y | n.a. | c.649A>T, p.(Lys217*)/ c.649A>T, p.(Lys217*) | Grammatikopoulos et al., 2016 [10] |
| 4/F | Caucasian/No | 21 wk | Jaundice, ascites, splenomegaly, GI bleeding; GGT 447 IU/L | Yes | 8 m—porto-portal bridging fibrosis, ductular reaction with ductal bile plugs. Hepatectomy specimen at 10 y—biliary cirrhosis, peripheral ductopenia. | Yes/10 y | 12 y | n.a. | c.890T>A, p.(Leu297*)/ c.890T>A, p.(Leu297*) | |
| 5/M | Arabic/Yes | 6 wk | Jaundice, GI bleeding; GGT 711 IU/L | n.a. | 8 wk—ductal plate malformation, cholestasis. Hepatectomy at 14 y—biliary cirrhosis, peripheral ductopenia. | Yes/14 y | 16 y | n.a. | c.757insG, p.(Ser253Argfs*4)/ c.757insG, p.(Ser253Argfs*4) | |
| 6/F | Caucasian/No | 4 wk | Jaundice, splenomegaly; GGT 210 IU/L | Yes | Hepatectomy at 15 y—porto-portal bridging fibrosis, peripheral ductopenia, ectasia and cystic dilatation of perihilar bile ducts. | Yes/15 y | Died at 17 y | n.a. | c.529dup, p.(Ile177Asnfs*20)/ c.890T>A, p.(Leu297*) | |
| 7/M | Caucasian/No | 6 wk | Jaundice, splenomegaly; GGT 962 IU/L | Yes | 9 wk—porto-portal bridging fibrosis, cholestasis, ductular proliferation with cholangiopathic features. 6 y—mild fibrosis, cholestasis, focal interlobular bile duct loss and cholangiopathic features in remaining bile ducts. | No | 6 y | n.a. | c.123_124del, p.(Ser42Glnfs*72)/ c.890T>A, p.(Leu297*) | |
| 8/M | Caucasian/No | 7 wk | Jaundice, splenomegaly; GGT 365 IU/L | Yes | 4 m—porto-portal bridging fibrosis, cholestasis, ductular proliferation and ductal bile plugs. Hepatectomy at 15 y—biliary cirrhosis, peripheral ductopaenia, ectasia and cystic dilatation of perihilar bile ducts. | Yes/15 y | 18 y | n.a. | c.123_124del, p.(Ser42Glnfs*72)/ c.123_124del, p.(Ser42Glnfs*72) | |
| 9/F | Caucasian/No | 1 wk | Jaundice; GGT 196 IU/L | Yes | 10 wk—ductal plate malformation, ductal bile plugs. 9 y—mild portal fibrosis, cholestasis, interlobular portal tract ductopaenia. Hepatectomy at 14 y—biliary cirrhosis with peripheral ductopaenia, ectasia and cystic dilatation of perihilar bile ducts. | Yes/14 y | 24 y | n.a. | c.529dup, p.(Ile177Asnfs*20)/ c.890T>A, p.(Leu297*) | |
| 10/M | n.a./Yes | n.a. | Jaundice, acholic stools | Yes | Liver histology for all 4 patients typical for NSC with early portal fibrosis, bile duct proliferation and tortuous bile ducts surrounded by fibrosis | Yes/14 y | 14 y | Renal malformation—left vesico-ureteral reflux with left ureteral duplication, without ureteral dilatation. Small left kidney with right kidney hypertrophy compensation. Mild intellectual disability. Renal insufficiency after liver transplantation. | c.51G>C, p.(Lys17Asn)/ c.51G>C, p.(Lys17Asn) | Girard et al., 2016 [9] |
| 11/M Sibling of 10 | n.a./Yes | n.a. | Yes/25 y | 9 y | Renal insufficiency after liver transplantation. | c.51G>C, p.(Lys17Asn)/ c.51G>C, p.(Lys17Asn) | ||||
| 12/F | n.a./Yes | n.a. | Yes/6 y | n.a. | No renal disease (imaging and function) at the age of 6 y. | c.426_557del; p.(Phe142_Arg186del)/ c.426_557del; p.(Phe142_Arg186del) | ||||
| 13/M Sibling of 12 | n.a./Yes | n.a. | Yes/3.5 y | n.a. | 4 mo—hyperechogenic left kidney at ultrasound and hypophosphatemia; normal renal function at 3 y. | c.426_557del; p.(Phe142_Arg186del)/ c.426_557del; p.(Phe142_Arg186del) | ||||
| 14/M | Chinese/n.a. | 2 mo | Jaundice; GGT 161-1 092 IU/L | Yes | n.a. | No | Diagnosis at 3 y 2 mo | bilateral hydronephrosis at 3 y 2 mo | c.529dup, p.(Ile177Asnfs*20)/ c.529dup, p.(Ile177Asnfs*20) | Li et al., 2018 [11] |
| 15/M | Chinese/n.a. | 9 mo | jaundice | Yes | n.a. | No | Diagnosis at 9 y 9 mo | hydrocephalus and left internal carotid artery aneurysms with vascular malformations diagnosed at 9 y 9 mo | c.529dup, p.(Ile177Asnfs*20)/ c.529dup, p.(Ile177Asnfs*20) | |
| 16/F | African-Caribbean/Yes | n.a. | n.a. | Yes | 13 y—13 years, 8 months with liver fibrosis, chronic liver failure 13 y—liver fibrosis, chronic liver failure; Hepatectomy—diffuse micronodular biliary cirrhosis, ductular proliferation | Yes (KLTx)/13 y | Nephronophthisis; Atrophic echogenic kidneys with decreased corticomedullary differentiation; Unilateral sensorineural deafness; brain imaging abnormalities | c.383C>G, p.(Ser128*)/ c.383C>G, p.(Ser128*) | Slater et al., 2019 [12] | |
| 17/M | Chinese/No | 1 wk | Jaundice; GGT 247 IU/L | n.a. | early portal fibrosis and bile duct proliferation; 3 y—cirrhosis | Yes/23 y | n.a. | Abnormal creatinine, no renal biopsy | c.705-2A>G, p.?/ c.923-283_ 1023+141del, p.? | Lin et al., 2020 [14] |
| 18/M Sibling of 17 | Chinese/No | 1 wk | Jaundice; GGT 102 IU/L | n.a. | n.a. | Yes/12 y | 18 y | Abnormal creatinine at 18 y | c.705-2A>G, p.?/ c.923-283_1023+141del, p.? | |
| 19/M Sibling of 17 and 18 | Chinese/No | 1 wk | Jaundice | n.a. | Clinical suspicion of biliary atresia; cirrhosis and liver failure at 8 m | No | Died at 8 m | n.a. | c.705-2A>G, p.?/ c.923-283_1023+141del, p.? | |
| 20/M | Caucasian/Yes | 1 wk | Jaundice, high GGT | No | Explanted liver—cirrhosis, hepatocellular and canalicular cholestasis, giant-cell change of hepatocytes | Yes/8 mo | 2 y | n.a. | c.294-2A>G, p.?/ c.294-2A>G, p.? | Vogel et al., 2020 [13] |
| 21/M | Turkish/Yes | 2 wk | Jaundice, acholic stools | Yes | Liver biopsy—bilirubin stasis, cholangiolytic changes, and septal fibrosis. | No | 3 y 7 m | 2 y—bilateral nephronophthisis; Psychomotor delay | c.367_368del, p.(Ser123Glnfs*9)/ c.367_368del, p.(Ser123Glnfs*9) | Syryn et al., 2021 [15] |
| 22/M | Syria/Yes | 3 mo | Jaundice, hepatosplenomegaly | Yes | Liver biopsy—biliary cirrhosis; Portal hypertension with GI bleeding | Yes/2y 10 mo | n.a. | Normal renal function; Psychomotor delay, microcephaly | c.73G>A p.(Gly25Arg)/ c.73G>A p.(Gly25Arg)/ | |
| 23/M | Turkish/Yes | 1 mo | Jaundice | n.a. | Liver biopsy (5 y)—congenital hepatic fibrosis | Yes (KLtx)/ 14.5 y | n.a. | Burkitt lymphoma at 11 y with renal failure development | n.a. | Duztas et al., 2022 [16] |
| 24/F | Turkish/Yes | 1 mo | Jaundice | n.a. | Liver biopsy (6 y)—cholestatic liver cirrhosis, ductopenia | Yes/6 y | n.a. | 6 y—enlargement of the right kidney with a cystic mass | c.656C>G, p.(Pro2219Arg)/ c.656C>G, p.(Pro2219Arg)/ | |
| 25/F | Chinese/No | 1 wk | Jaundice, elevated GGT | Yes | Giant cell changes of hepatocytes, bile plugs in hepatocytes and capillary bile ducts; ductular proliferation, cholestatic cirrhosis | No | n.a. | n.a. | c.1024-1G>T, p.?/ c.544G>A, p.(Gly182Arg) | Wei et al., 2023 [17] |
| 26/F | Chinese/No | 1 wk | Jaundice, elevated GGT | Yes | Cholestasis, liver fibrosis (stage 3), ductular proliferation, giant cell changes of hepatocytes | No | n.a. | n.a. | c.1024-1G>T, p.?/ c.544G>A, p.(Gly182Arg) | |
| 27/M | Chinese/No | 1 wk | Jaundice, elevated GGT | Yes | No liver biopsy | No | n.a. | n.a. | c.529dup, p.(Ile177Asnfs*20)/ c.529dup, p.(Ile177Asnfs*20) | |
| 28/M | Chinese/No | 1 wk | Jaundice, elevated GGT | No | No liver biopsy | No | n.a. | n.a. | c.529dup, p.(Ile177Asnfs*20)/ c.529dup, p.(Ile177Asnfs*20) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipiński, P.; Ciara, E.; Jurkiewicz, D.; Mekrouda, M.; Cielecka-Kuszyk, J.; Jurkiewicz, E.; Płoski, R.; Pawłowska, J.; Jankowska, I. DCDC2-Related Ciliopathy: Report of Six Polish Patients, Novel DCDC2 Variant, and Literature Review of Reported Cases. Diagnostics 2023, 13, 1917. https://doi.org/10.3390/diagnostics13111917
Lipiński P, Ciara E, Jurkiewicz D, Mekrouda M, Cielecka-Kuszyk J, Jurkiewicz E, Płoski R, Pawłowska J, Jankowska I. DCDC2-Related Ciliopathy: Report of Six Polish Patients, Novel DCDC2 Variant, and Literature Review of Reported Cases. Diagnostics. 2023; 13(11):1917. https://doi.org/10.3390/diagnostics13111917
Chicago/Turabian StyleLipiński, Patryk, Elżbieta Ciara, Dorota Jurkiewicz, Magda Mekrouda, Joanna Cielecka-Kuszyk, Elżbieta Jurkiewicz, Rafał Płoski, Joanna Pawłowska, and Irena Jankowska. 2023. "DCDC2-Related Ciliopathy: Report of Six Polish Patients, Novel DCDC2 Variant, and Literature Review of Reported Cases" Diagnostics 13, no. 11: 1917. https://doi.org/10.3390/diagnostics13111917
APA StyleLipiński, P., Ciara, E., Jurkiewicz, D., Mekrouda, M., Cielecka-Kuszyk, J., Jurkiewicz, E., Płoski, R., Pawłowska, J., & Jankowska, I. (2023). DCDC2-Related Ciliopathy: Report of Six Polish Patients, Novel DCDC2 Variant, and Literature Review of Reported Cases. Diagnostics, 13(11), 1917. https://doi.org/10.3390/diagnostics13111917