
Machine Learning of Raman Spectroscopy Data for Classifying Cancers: A Review of the Recent Literature

 , , ,
, , ,
Abstract
:1. Introduction
Assessing Model Performance
2. Materials and Methods
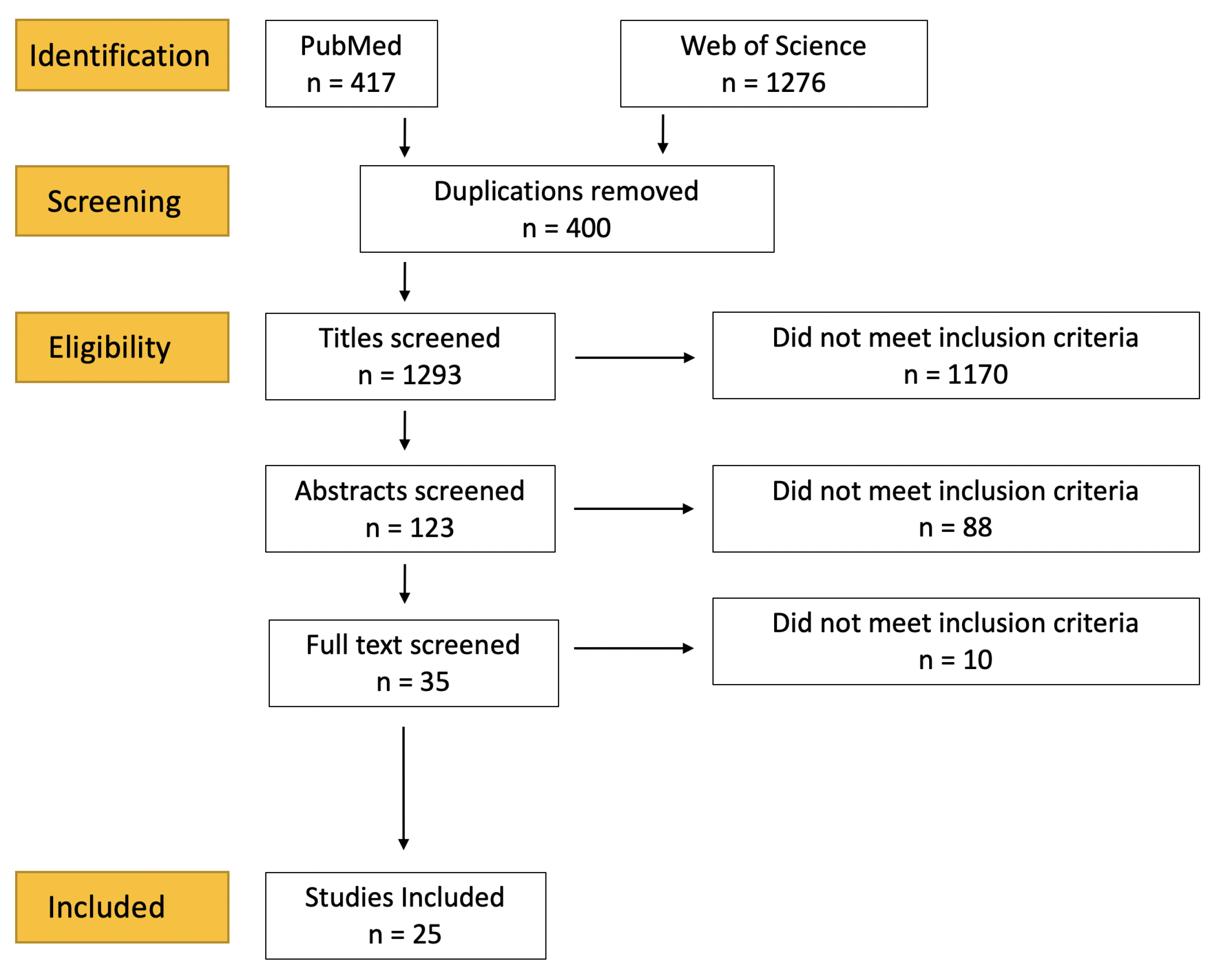
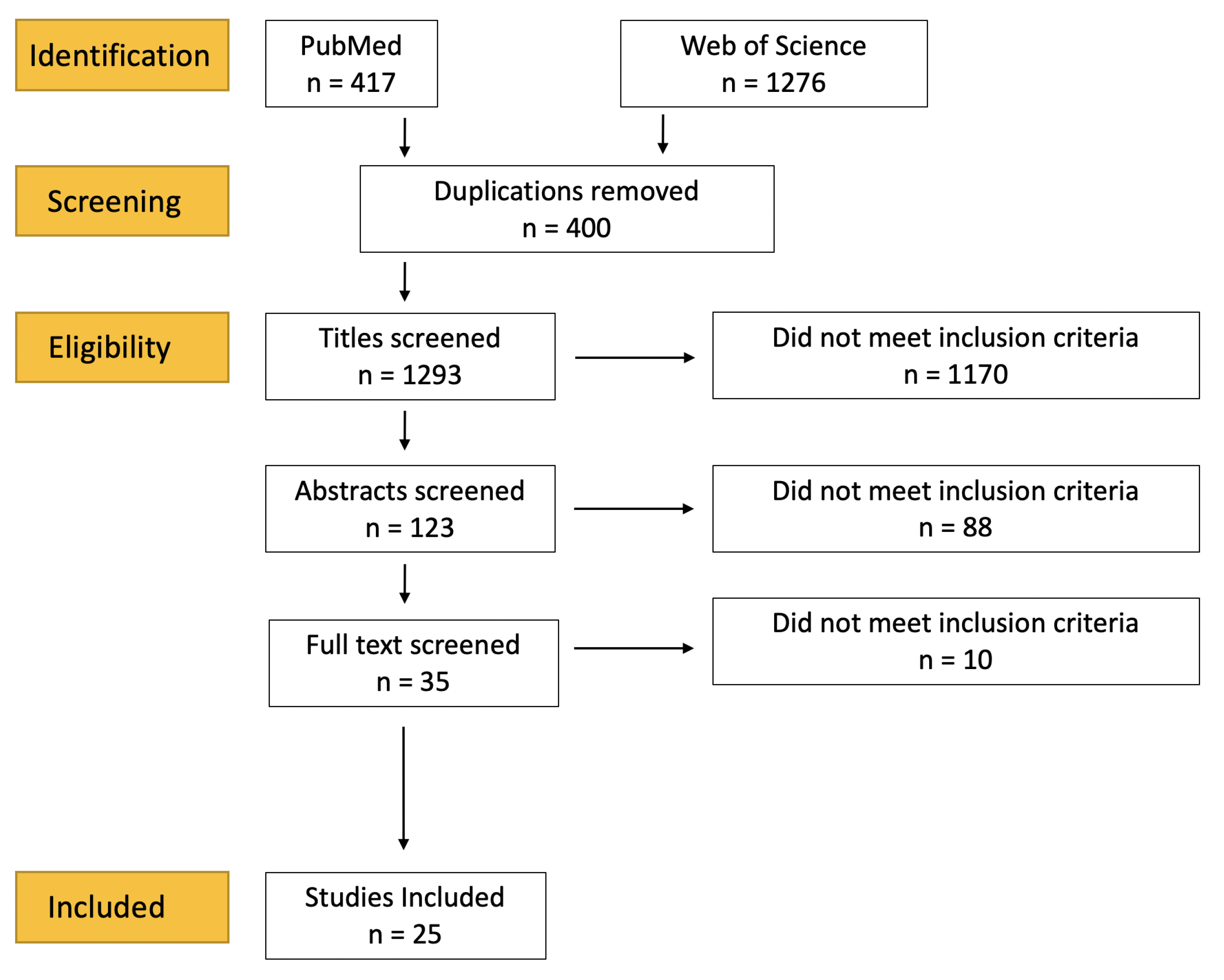
2.1. Literature Search
2.2. Data Collection
3. Results
3.1. Oral and Nasopharangeal Cancers
3.2. Lung Cancers
3.3. Brain Cancers
3.4. Breast Cancers
3.5. Prostate Cancers
3.6. Gastrointestinal Cancers
3.7. Skin Cancers
3.8. Gynaecological Cancers
3.9. Other Cancers
4. Discussion
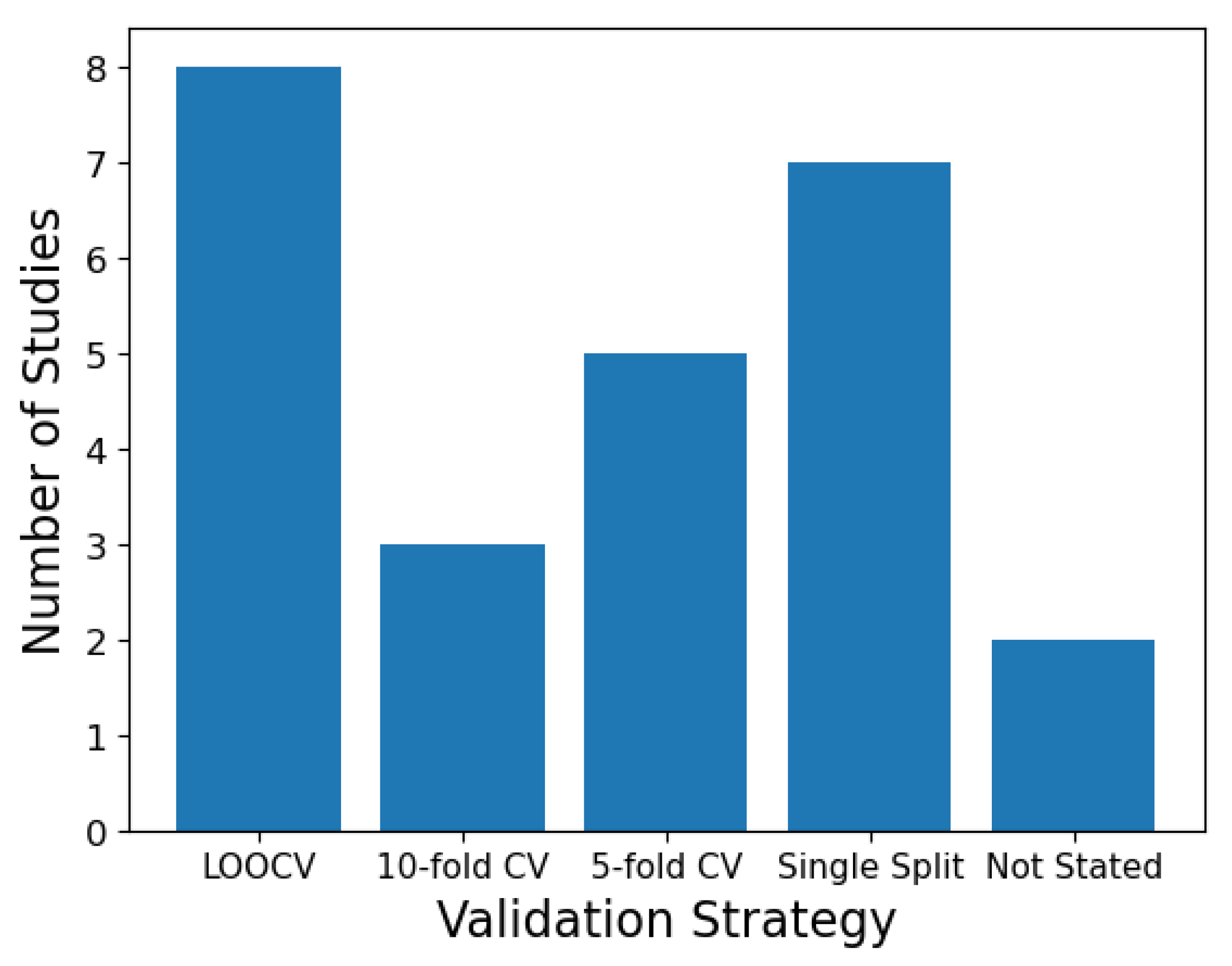
4.1. Validation Strategies
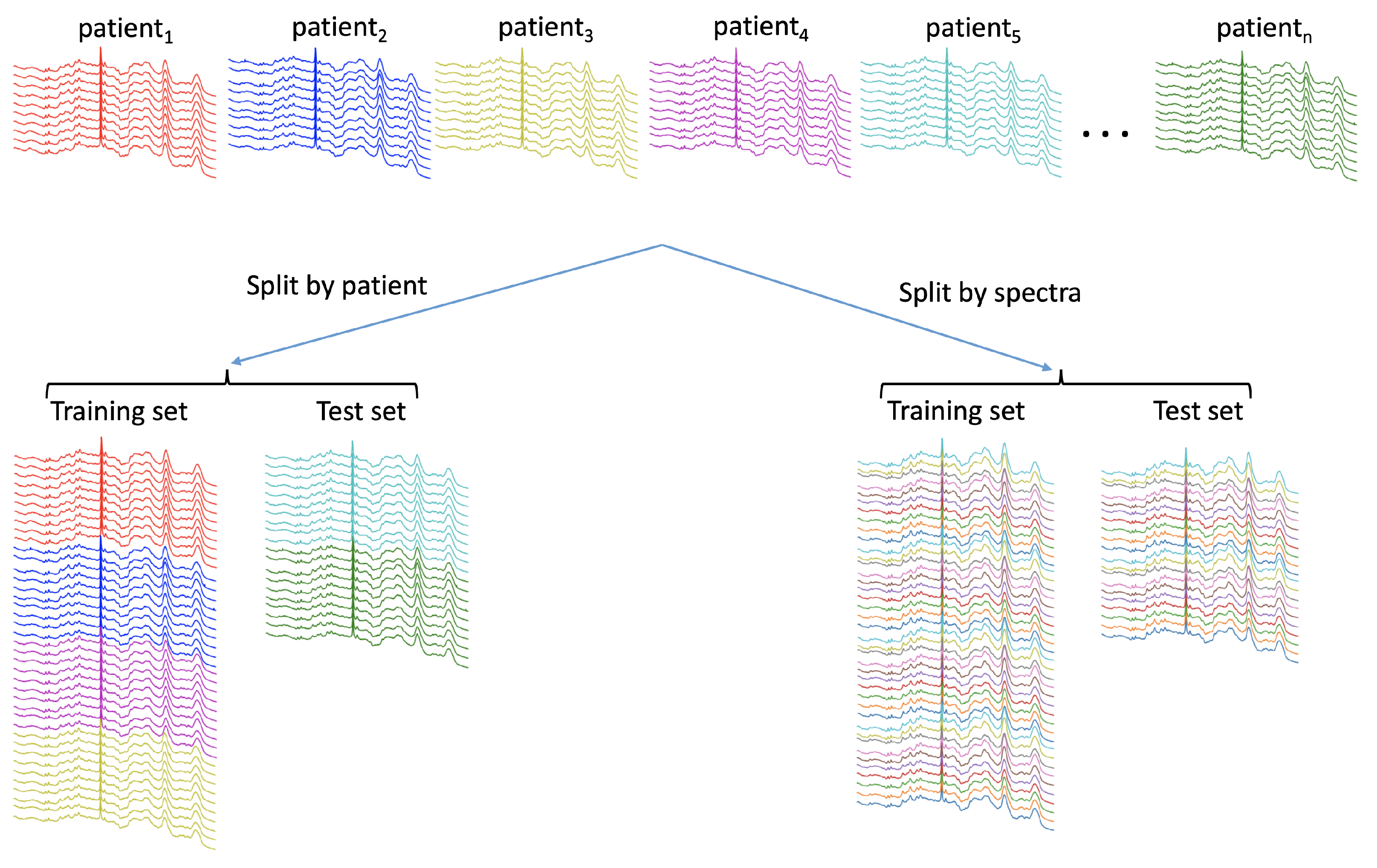
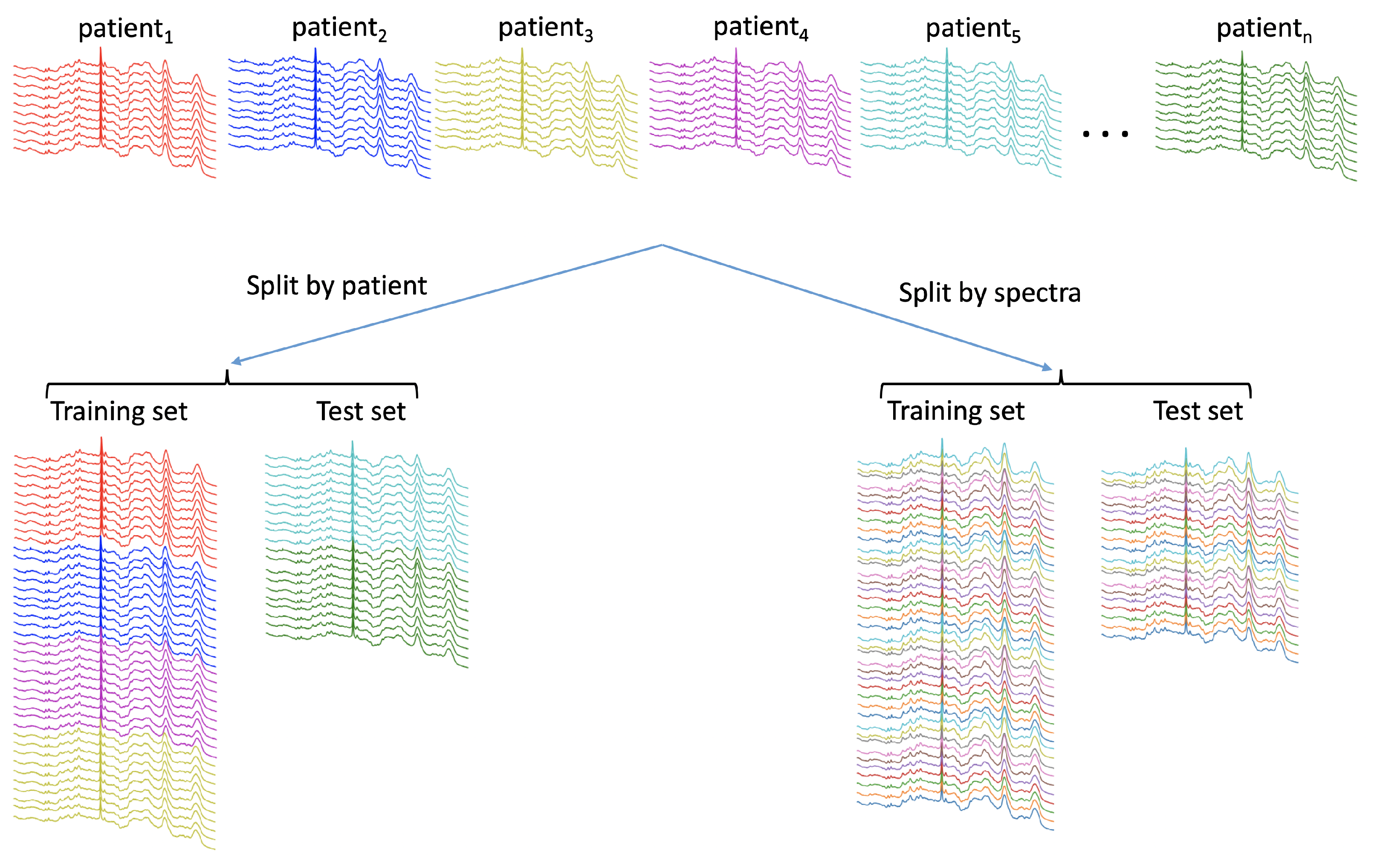
4.1.1. Partitioning Data with a Hierarchical Structure
4.1.2. Paired Sampling
4.1.3. Sample Representativeness and Label Noise
4.2. Data Augmentation
4.3. Pre-Processing
4.4. Traditional versus Deep Machine Learning
4.5. Model Transparency and Interpretability
4.6. Recommendations
- When data are split into training, validation and test sets, the level of the split should be conducted at the highest level (usually the subject, but this may be coincident with the sample). The chosen level should be explicitly stated.
- If model hyper-parameters are explored, including pre-processing, these should be selected based on a validation set and once selected only then tested against the test.
- All pre-processing and data augmentation techniques should be described in sufficient detail to allow for replication. While there is doubt regarding the need for pre-processing with CNNs, it would be beneficial for studies to include results from both raw and pre-processed data.
- All the hyper-parameters used in the model should be stated and the hyper-parameter search strategy should be reported.
- Where possible, consensus pathology should be used to confirm class labels.
- Where possible, attempts should be made to relate the results to the underlying biochemistry by interrogating model outputs, in addition to the traditional methods of comparing average spectra per class, peak comparisons and obtaining difference spectra.
- Resampling validation strategies should be used, such as k-fold and/or repeated CV. As these methods provide many generalisation estimates, standard deviations of performance metrics should be reported.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RS | Raman Spectroscopy |
| NIR | Near-Infrared |
| PCA | Principle Component Analysis |
| LDA | Linear Discriminant Analysis |
| QDA | Quadratic Discriminant Analysis |
| PLS | Partial Least Squares |
| SVM | Support Vector Machine |
| ANN | Artificial Neural Network |
| CNN | Convolutional Neural Network |
| KNN | K-Nearest Neighbours |
| RF | Random Forest |
| GB | Gradient Boost |
| LSTM | Long Short Term Memory |
| RNN | Recurrent Neural Network |
| RBF | Radial Basis Function |
| CV | Cross Validation |
| LOOCV | Leave One Out Cross Validation |
| ATR-FTIR | attenuated total reflection-Fourier transform infrared |
| ML | Machine Learning |
| NNLS | Non-Negative Least Squares |
| EMSC | Extended Multiplicative Scatter Correction |
| GA | Genetic Algorithm |
| NPC | Nasopharyngeal Carcinoma |
References
- Santos, I.P.; Barroso, E.M.; Schut, T.C.B.; Caspers, P.J.; van Lanschot, C.G.F.; Choi, D.H.; van der Kamp, M.F.; Smits, R.W.H.; van Doorn, R.; Verdijk, R.M.; et al. Raman spectroscopy for cancer detection and cancer surgery guidance: Translation to the clinics. Analyst 2017, 142, 3025–3047. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Hudecek, J.; Mulder, L.; van Seijen, M.; Almekinders, M.M.; Alexov, S.; Kovács, A.; Ryska, A.; Varga, Z.; Navarro, F.J.A.; et al. Prognostic value of histopathological DCIS features in a large-scale international interrater reliability study. Breast Cancer Res. Treat. 2020, 183, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Mehlum, C.S.; Larsen, S.R.; Kiss, K.; Groentved, A.M.; Kjaergaard, T.; Möller, S.; Godballe, C. Laryngeal precursor lesions: Interrater and intrarater reliability of histopathological assessment. Laryngoscope 2018, 128, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Barnard, M.E.; Pyden, A.; Rice, M.S.; Linares, M.; Tworoger, S.S.; Howitt, B.E.; Meserve, E.E.; Hecht, J.L. Inter-pathologist and pathology report agreement for ovarian tumor characteristics in the Nurses’ Health Studies. Gynecol. Oncol. 2018, 150, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, N. Cancer survival data emphasise importance of early diagnosis. BMJ 2019, 364, 408. [Google Scholar] [CrossRef] [PubMed]
- Picot, F.; Daoust, F.; Sheehy, G.; Dallaire, F.; Chaikho, L.; Bégin, T.; Kadoury, S.; Leblond, F. Data consistency and classification model transferability across biomedical Raman spectroscopy systems. Transl. Biophotonics 2021, 3, e202000019. [Google Scholar] [CrossRef]
- Esteva, A.; Robicquet, A.; Ramsundar, B.; Kuleshov, V.; DePristo, M.; Chou, K.; Cui, C.; Corrado, G.; Thrun, S.; Dean, J. A guide to deep learning in healthcare. Nat. Med. 2019, 25, 24–29. [Google Scholar] [CrossRef]
- Pradhan, P.; Guo, S.; Ryabchykov, O.; Popp, J.; Bocklitz, T.W. Deep learning a boon for biophotonics? J. Biophotonics 2020, 13, e201960186. [Google Scholar] [CrossRef] [Green Version]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology—New tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef]
- Rajkomar, A.D.J.; Kohane, I. Machine learning in medicine. N. Engl. J. Med. 2019, 380, 1347–1358. [Google Scholar] [CrossRef]
- Roberts, M.; Driggs, D.; Thorpe, M.; Gilbey, J.Y.M.; Ursprung, S.; Aviles-Rivero, A.; Etmann, C.; McCague, C.; Beer, L.; Weir-McCall, J. Common pitfalls and recommendations for using machine learning to detect and prognosticate for COVID-19 using chest radiographs and CT scans. Nat. Mach. Intell. 2021, 3, 199–217. [Google Scholar] [CrossRef]
- Baker, M.J.; Byrne, H.J.; Chalmers, J.; Gardner, P.; Goodacre, R.; Henderson, A.; Kazarian, S.G.; Martin, F.L.; Moger, J.; Stone, N.; et al. Clinical applications of infrared and Raman spectroscopy: State of play and future challenges. Analyst 2018, 143, 1735–1757. [Google Scholar] [CrossRef]
- Cawley, G.C.; Talbot, N.L. On over-fitting in model selection and subsequent selection bias in performance evaluation. J. Mach. Learn. Res. 2010, 11, 2079–2107. [Google Scholar]
- Molinaro, A.M.; Simon, R.; Pfeiffer, R.M. Prediction error estimation: A comparison of resampling methods. Bioinformatics 2005, 21, 3301–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.; McKenzie, J.; Bossuyt, P.; Boutron, I.; Hoffmann, T.; Mulrow, C.; Shamseer, L.; Tetzlaff, J.; Akl, E.; Brennan, S.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, 160. [Google Scholar] [CrossRef] [PubMed]
- Aubertin, K.; Trinh, V.Q.; Jermyn, M.; Baksic, P.; Grosset, A.A.; Desroches, J.; St-Arnaud, K.; Birlea, M.; Vladoiu, M.C.; Latour, M.; et al. Mesoscopic characterization of prostate cancer using Raman spectroscopy: Potential for diagnostics and therapeutics. BJU Int. 2018, 122, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Baria, E.; Cicchi, R.; Malentacchi, F.; Mancini, I.; Pinzani, P.; Pazzagli, M.; Pavone, F.S. Supervised learning methods for the recognition of melanoma cell lines through the analysis of their Raman spectra. J. Biophotonics 2021, 14, 202000365. [Google Scholar] [CrossRef]
- Bury, D.; Faust, G.; Paraskevaidi, M.; Ashton, K.M.; Dawson, T.P.; Martin, F.L. Phenotyping metastatic brain tumors applying spectrochemical analyses: Segregation of different cancer types. Anal. Lett. 2019, 52, 575–587. [Google Scholar] [CrossRef]
- Chen, F.; Sun, C.; Yue, Z.; Zhang, Y.; Xu, W.; Shabbir, S.; Zou, L.; Lu, W.; Wang, W.; Xie, Z.; et al. Screening ovarian cancers with Raman spectroscopy of blood plasma coupled with machine learning data processing. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 265, 120355. [Google Scholar] [CrossRef]
- Chen, C.; Wu, W.; Chen, C.; Chen, F.; Dong, X.; Ma, M.; Yan, Z.; Lv, X.; Ma, Y.; Zhu, M. Rapid diagnosis of lung cancer and glioma based on serum Raman spectroscopy combined with deep learning. J. Raman Spectrosc. 2021, 52, 1798–1809. [Google Scholar] [CrossRef]
- Daniel, A.; Prakasarao, A.; Ganesan, S. Near-infrared Raman spectroscopy for estimating biochemical changes associated with different pathological conditions of cervix. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 409–416. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wu, X.; Zhou, J.; Chen, Y.; Ye, J. Raman optical identification of renal cell carcinoma via machine learning. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 252, 119520. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Uragami, N.; Miyazaki, T.; Yang, W.; Issha, K.; Matsuo, K.; Kimura, S.; Arai, Y.; Tokunaga, H.; Okada, S.; et al. Highly accurate colorectal cancer prediction model based on Raman spectroscopy using patient serum. World J. Gastrointest. Oncol. 2020, 12, 1311. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.; Sharma, L.C.T.; Huang, S.; Chang, L.; Wu, S.; Chow, L. Raman spectroscopy analysis for optical diagnosis of oral cancer detection. J. Clin. Med. 2019, 8, 1313. [Google Scholar] [CrossRef] [PubMed]
- Koya, S.; Brusatori, M.; Yurgelevic, S.; Huang, C.; Werner, C.; Kast, R.; Shanley, J.; Sherman, M.; Honn, K.; Maddipati, K.; et al. Accurate identification of breast cancer margins in microenvironments of ex-vivo basal and luminal breast cancer tissues using Raman spectroscopy. Prostaglandins Other Lipid Mediat. 2020, 151, 106475. [Google Scholar] [CrossRef]
- Lee, W.; Lenferink, A.; Otto, C.; Offerhaus, H. Classifying Raman spectra of extracellular vesicles based on convolutional neural networks for prostate cancer detection. J. Raman Spectrosc. 2020, 51, 293–300. [Google Scholar] [CrossRef]
- Ma, D.; Shang, L.; Tang, J.; Bao, Y.; Fu, J.; Yin, J. Classifying breast cancer tissue by Raman spectroscopy with one-dimensional convolutional neural network. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 256, 119732. [Google Scholar] [CrossRef]
- Mehta, K.; Atak, A.; Sahu, A.; Srivastava, S. An early investigative serum Raman spectroscopy study of meningioma. Analyst 2018, 143, 1916–1923. [Google Scholar] [CrossRef]
- Qi, Y.; Yang, L.; Liu, B.; Liu, L.; Liu, Y.; Zheng, Q.; Liu, D.; Luo, J. Highly accurate diagnosis of lung adenocarcinoma and squamous cell carcinoma tissues by deep learning. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 265, 120400. [Google Scholar] [CrossRef]
- Riva, M.; Sciortino, T.; Secoli, R.; D’Amico, E.; Moccia, S.; Fernandes, B.; Conti Nibali, M.; Gay, L.; Rossi, M.; De Momi, E.; et al. Glioma biopsies Classification Using Raman Spectroscopy and Machine Learning Models on Fresh Tissue Samples. Cancers 2021, 13, 1073. [Google Scholar] [CrossRef]
- Santos, I.P.; van Doorn, R.; Caspers, P.J.; Schut, T.C.B.; Barroso, E.M.; Nijsten, T.E.; Hegt, V.N.; Koljenović, S.; Puppels, G.J. Improving clinical diagnosis of early-stage cutaneous melanoma based on Raman spectroscopy. Br. J. Cancer 2018, 119, 1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciortino, T.; Secoli, R.; d’Amico, E.; Moccia, S.; Conti Nibali, M.; Gay, L.; Rossi, M.; Pecco, N.; Castellano, A.; De Momi, E.; et al. Raman Spectroscopy and Machine Learning for IDH Genotyping of Unprocessed Glioma Biopsies. Cancers 2021, 13, 4196. [Google Scholar] [CrossRef] [PubMed]
- Serzhantov, K.A.; Myakinin, O.O.; Lisovskaya, M.G.; Bratchenko, I.A.; Moryatov, A.A.; Kozlov, S.V.; Zakharov, V.P. Comparison testing of machine learning algorithms separability on Raman spectra of skin cancer. SPIE 2020, 11359, 1135906. [Google Scholar]
- Shu, C.; Yan, H.; Zheng, W.; Lin, K.; James, A.; Selvarajan, S.; Lim, C.; Huang, Z. Deep Learning-Guided Fiberoptic Raman Spectroscopy Enables Real-Time In Vivo Diagnosis and Assessment of Nasopharyngeal Carcinoma and Post-treatment Efficacy during Endoscopy. Anal. Chem. 2021, 93, 10898–10906. [Google Scholar] [CrossRef]
- Wu, X.; Li, S.; Xu, Q.; Yan, X.; Fu, Q.; Fu, X.; Fang, X.; Zhang, Y. Rapid and accurate identification of colon cancer by Raman spectroscopy coupled with convolutional neural networks. Jpn. J. Appl. Phys. 2021, 60, 067001. [Google Scholar] [CrossRef]
- Xia, J.; Zhu, L.; Yu, M.; Zhang, T.; Zhu, Z.; Lou, X.; Sun, G.; Dong, M. Analysis and classification of oral tongue squamous cell carcinoma based on Raman spectroscopy and convolutional neural networks. J. Mod. Opt. 2020, 67, 481–489. [Google Scholar] [CrossRef]
- Yan, H.; Yu, M.; Xia, J.; Zhu, L.; Zhang, T.; Zhu, Z.; Sun, G. Diverse Region-Based CNN for Tongue Squamous Cell Carcinoma Classification With Raman Spectroscopy. IEEE Access 2020, 8, 127313–127328. [Google Scholar] [CrossRef]
- Yu, M.; Yan, H.; Xia, J.; Zhu, L.; Zhang, T.; Zhu, Z.; Lou, X.; Sun, G.; Dong, M. Deep convolutional neural networks for tongue squamous cell carcinoma classification using Raman spectroscopy. Photodiagn. Photodyn. Ther. 2019, 26, 430–435. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Peng, D.; Yi, X.; He, S.; Liu, F.; Zheng, X.; Huang, W.; Zhao, L.; Huang, X. Raman spectroscopy and machine learning for the classification of breast cancers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 264, 120300. [Google Scholar] [CrossRef]
- Zuvela, P.; Lin, K.; Shu, C.; Zheng, W.; Lim, C.; Huang, Z. Fiber-optic Raman spectroscopy with nature-inspired genetic algorithms enhances real-time in vivo detection and diagnosis of nasopharyngeal carcinoma. Anal. Chem. 2019, 91, 8101–8108. [Google Scholar] [CrossRef]
- Bengio, Y.; Grandvalet, Y. No unbiased estimator of the variance of k-fold cross-validation. J. Mach. Learn. Res. 2004, 5, 1089–1105. [Google Scholar]
- Beleites, C.; Baumgartner, R.; Bowman, C.; Somorjai, R.; Steiner, G.; Salzer, R.; Sowa, M.G. Variance reduction in estimating classification error using sparse datasets. Chemom. Intell. Lab. Syst. 2005, 79, 91–100. [Google Scholar] [CrossRef]
- Rácz, A.; Bajusz, D.; Héberger, K. Modelling methods and cross-validation variants in QSAR: A multi-level analysis. SAR QSAR Environ. Res. 2018, 29, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Bocklitz, T.; Neugebauer, U.; Popp, J. Common mistakes in cross-validating classification models. Anal. Methods 2017, 9, 4410–4417. [Google Scholar] [CrossRef]
- Paidi, S.K.; Pandey, R.; Barman, I. Emerging trends in biomedical imaging and disease diagnosis using Raman spectroscopy. In Molecular and Laser Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 623–652. [Google Scholar]
- Frénay, B.; Kabán, A. A comprehensive introduction to label noise. In ESANN; Citeseer: Bruges, Belgium, 2014. [Google Scholar]
- Santos, I.P.; Caspers, P.J.; Bakker Schut, T.C.; van Doorn, R.; Noordhoek Hegt, V.; Koljenović, S.; Puppels, G.J. Raman spectroscopic characterization of melanoma and benign melanocytic lesions suspected of melanoma using high-wavenumber Raman spectroscopy. Anal. Chem. 2016, 88, 7683–7688. [Google Scholar] [CrossRef]
- Svensson, C.M.; Hübler, R.; Figge, M.T. Automated classification of circulating tumor cells and the impact of interobsever variability on classifier training and performance. J. Immunol. Res. 2015, 2015, 573165. [Google Scholar] [CrossRef] [Green Version]
- Karimi, D.; Dou, H.; Warfield, S.K.; Gholipour, A. Deep learning with noisy labels: Exploring techniques and remedies in medical image analysis. Med. Image Anal. 2020, 65, 101759. [Google Scholar] [CrossRef]
- Shorten, C.; Khoshgoftaar, T.M. A survey on image data augmentation for deep learning. J. Big Data 2019, 6, 1–48. [Google Scholar] [CrossRef]
- Fang, X.; Zeng, Q.; Yan, X.; Zhao, Z.; Chen, N.; Deng, Q.; Zhu, M.; Zhang, Y.; Li, S. Fast discrimination of tumor and blood cells by label-free surface-enhanced Raman scattering spectra and deep learning. J. Appl. Phys. 2021, 129, 123103. [Google Scholar] [CrossRef]
- Perez, F.; Vasconcelos, C.; Avila, S.; Valle, E. Data augmentation for skin lesion analysis. In OR 2.0 Context-Aware Operating Theaters, Computer Assisted Robotic Endoscopy, Clinical Image-Based Procedures, and Skin Image Analysis; Springer: Cham, Switzerland, 2018; pp. 303–311. [Google Scholar]
- Bocklitz, T.; Walter, A.; Hartmann, K.; Rösch, P.; Popp, J. How to pre-process Raman spectra for reliable and stable models? Anal. Chim. Acta 2011, 704, 47–56. [Google Scholar] [CrossRef]
- Vollmer, S.; Mateen, B.; Bohner, G.; Király, F.; Ghani, R.; Jonsson, P.; Cumbers, S.; Jonas, A.; McAllister, K.; Myles, P.; et al. Machine learning and AI research for patient benefit: 20 critical questions on transparency, replicability, ethics and effectiveness. arXiv 2018, arXiv:1812.10404. [Google Scholar]
- Nagendran, M.; Chen, Y.; Lovejoy, C.A.; Gordon, A.C.; Komorowski, M.; Harvey, H.; Topol, E.J.; Ioannidis, J.P.; Collins, G.S.; Maruthappu, M. Artificial intelligence versus clinicians: Systematic review of design, reporting standards, and claims of deep learning studies. BMJ 2020, 368, 689. [Google Scholar] [CrossRef] [Green Version]
- Zech, J.R.; Badgeley, M.A.; Liu, M.; Costa, A.B.; Titano, J.J.; Oermann, E.K. Variable generalization performance of a deep learning model to detect pneumonia in chest radiographs: A cross-sectional study. PLoS Med. 2018, 15, e1002683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Heinke, R.; Stöckel, S.; Rösch, P.; Bocklitz, T.; Popp, J. Towards an improvement of model transferability for Raman spectroscopy in biological applications. Vib. Spectrosc. 2017, 91, 111–118. [Google Scholar] [CrossRef]
- Guo, S.; Popp, J.; Bocklitz, T. Chemometric analysis in Raman spectroscopy from experimental design to machine learning–based modeling. Nat. Protoc. 2021, 16, 5426–5459. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Authors/Year | Pathology Sample Type | Model | Validation Strategy | Number of Subjects/ Samples | Number of Spectra | Level of Split | Number of Classes | Accuracy (Sensitivity/ Specificity) |
|---|---|---|---|---|---|---|---|---|
| Aubertin et al., 2018 [16] | Prostate Cancer (tissue) | ANN | LOOCV | 32 subjects/ samples | 928 | Not Stated | 2 | 86% (87%/86%) |
| Baria et al., 2020 [17] | Skin Cancer (cell lines) | PCA-ANN | 5-fold CV | Not Stated | 150 | Not Stated | 3 | 96.7% |
| Bury et al., 2019 [18] | Brain Metastases (tissue) | PCA-LDA | Not Stated | 21 subjects | 525 | Not Stated | 2 | 80.2% |
| Chen et al., 2022 [19] | Ovarian Cancer (plasma) | ANN ensemble | Outer fold–single 66/33 Inner fold–5-fold CV | 174 subjects | 870 | Spectra | 2 | 94.8% (95%/95%) |
| Chen et al., 2021 [20] | Lung cancer & glioma (tissue) | CNN | 5-fold CV | 104 subjects/ samples | 520 (2700 post augmentation) | Subject | 2 | 99% (all pairwise comparisons > 95% |
| Daniel et al., 2019 [21] | Cervical Cancer (tissue) | PCA-ANN | Single 70/30 | 245 samples | Not Stated | Not Stated | 3 | 99.0% (87%/86%) |
| He et al., 2021 [22] | Renal Cancer | SVM | LOOCV | 77 subjects/ samples | 4860 | Subject | 3 | 92.89% |
| Ito et al., 2020 [23] | Colon Cancer (serum) | Boosted Tree | Not Stated | 184 subjects/ samples | 3 spectra per subject. Average used | N/A | 2 | 100% |
| Jeng et al., 2019 [24] | Oral Cancer (tissue) | PCA-QDA | k-fold CV and LOOCV | 80 subjects/ samples | 400 | Sample | 2 | 82% (84%/75%) |
| Koya et al., 2020 [25] | Breast Cancer (tissue) | CNN | Single split 60/20/20 | 88 subjects/ samples | 34,505 | Spectra | 2 | 90% (89%—precision, 89%—recall) |
| Lee et al., 2020 [26] | Prostate Cancer (cell lines) | CNN | Single split 70/15/15 | 1 sample per class, 4 classes | 300 (1200 post augmentation) | Spectra | 4 | 97% |
| Ma et al., 2021 [27] | Breast Cancer (tissue) | CNN | 10-fold CV | 20 subjects, 40 samples | 600 (5000 post augmentation) | Not Stated | 2 | 92% (98%/86%) |
| Mehta et al., 2018 [28] | Brain Meningioma (serum) | PCA-LDA | LOOCV + independent test set | 20 subjects, 70 samples | ~8 spectra per subject. Average used | N/A | 2 | 86% |
| Qi et al., 2022 [29] | Lung Cancer (tissue) | CNN | 10-fold CV | 77 subjects/ samples | 15 spectra per sample | Spectra | 2 | 98% (97%/99%) |
| Riva et al., 2021 [30] | Glioma (tissue) | GB | LOOCV | 63 subjects/ samples | 3450 | Subject | 2 | 83% (82%—precision, 82%—recall) |
| Santos et al., 2018 [31] | Skin (tissue) | PCA-LDA | Single split 60/40 | 128 samples | 9–19 spectra per sample | Sample | 2 | 62.5% |
| Sciortino et al., 2021 [32] | Glioma (tissue) | SVM | LOOCV | 38 subjects/ samples | 2073 | Subject | 2 | 87% |
| Serzhantov et al., 2020 [33] | Skin (tissue) | Gradient with soft voting | Single split 50/50, 1000 repeats | 139 subjects | 556 | Not Stated | 2 | 91% (93%/88%) |
| Shu et al., 2021 [34] | Nasopharyngeal Cancer (in vivo tissue) | CNN | 10-fold Venetian Blind CV | 418 subjects, 888 samples | 15,354 (Augmented, quantity not specified) | Sample | 2 | 84% (99%/66%) |
| Wu et al., 2021 [35] | Colon Cancer (tissue) | CNN | LOOCV | 45 subjects/ samples | 233 (2420 post augmentation) | Spectra AND Subject | 3 | 94%—by spectra, 81%—by subject |
| Xia et al., 2021 [36] | Tongue Cancer (tissue) | CNN-SVM | 5-fold CV | 12 subjects, 24 samples | At least 216 | Not Stated | 2 | 99.5% (100%/100%) |
| Yan et al., 2021 [37] | Tongue Cancer (tissue) | CNN ensemble | 5-fold CV | 22 subjects, 44 samples | 2004 | Not Stated | 2 | 99% (99%/98%) |
| Yu et al., 2021 [38] | Tongue Cancer (tissue) | CNN | 5-fold CV | 12 subjects, 24 samples | 1440 | Not Stated | 2 | 97% (99%/94%) |
| Zhang et al., 2021 [39] | Breast Cancer (cell lines) | PCA-SVM | Single split | 6 cell line 900 cells | 4500 | Not Stated | 2 | 99.0% (100%/96%) |
| Zuvela et al., 2019 [40] | Nasopharyngeal Cancer (in vivo tissue) | GA- PLS-LDA | LOOCV | 62 subjects, 113 samples | 2126 | Sample | 2 | 98% (93%/100%) |
| Study | Deep Model | Traditional Model | Data Subsets |
|---|---|---|---|
| Baria et al., 2020 [17] | 96.0% (PCA-ANN) | 98.0% (PCA-LDA) | SK-MEL-2 (Cell lines) |
| 96.0% | 90.0% | SK-MEL-28 | |
| 98.0% | 90.0% | MW-266-4 | |
| 96.7% | 92.7% | All | |
| Daniel et al., 2019 [21] | 99.0% (PCA-ANN) | 98.0% (PCA-LDA) | |
| He et al., 2021 [22] | 92.3% (ANN) | 92.9% (SVM) | |
| Lee et al., 2020 [26] | 90.9% (CNN) | 78.3% (PCA-QDA) | Processed, whole spectra |
| 90.2% | 95.0% | Processed, fingerprint | |
| 91.2% | 86.7% | Processed, high wavenumber | |
| 95.2% | 68.3% | Unprocessed, whole spectra | |
| 96.6% | 61.7% | Unprocessed, fingerprint | |
| 93.1% | 60.0% | Unprocessed, high wavenumber | |
| Ma et al., 2021 [27] | 92.0% (ANN) | 86.5% (SVM) | |
| Qi et al., 2022 [29] | 97.7% (ANN) | 86.6% (PCA-LDA) | Adenomcarcinoma |
| 96.1% | 82.1% | Squamous cell carcinoma | |
| Shu et al., 2021 [34] | 82.1% (CNN) | 73.6% (PLS-LDA) | All data |
| 84.4% | 83.7% | NPC vs. control | |
| 82.1% | 68.4% | NPC vs. post-treatment | |
| Wu et al., 2021 [35] | 81.3% (CNN) | 52.7% (KNN) | Processed data |
| 75.0% | 42.0% (SVM) | Unprocessed data | |
| Xia et al., 2021 [36] | 99.6% (CNN-SVM) | 95.4% (PCA-SVM) | |
| Yan et al., 2021 [37] | 98.8% (Ensemble CNN) | 88.5% (PCA-SVM) | |
| Yu et al., 2021 [38] | 96.9% (CNN) | 88.5% (SVM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blake, N.; Gaifulina, R.; Griffin, L.D.; Bell, I.M.; Thomas, G.M.H. Machine Learning of Raman Spectroscopy Data for Classifying Cancers: A Review of the Recent Literature. Diagnostics 2022, 12, 1491. https://doi.org/10.3390/diagnostics12061491
Blake N, Gaifulina R, Griffin LD, Bell IM, Thomas GMH. Machine Learning of Raman Spectroscopy Data for Classifying Cancers: A Review of the Recent Literature. Diagnostics. 2022; 12(6):1491. https://doi.org/10.3390/diagnostics12061491
Chicago/Turabian StyleBlake, Nathan, Riana Gaifulina, Lewis D. Griffin, Ian M. Bell, and Geraint M. H. Thomas. 2022. "Machine Learning of Raman Spectroscopy Data for Classifying Cancers: A Review of the Recent Literature" Diagnostics 12, no. 6: 1491. https://doi.org/10.3390/diagnostics12061491
APA StyleBlake, N., Gaifulina, R., Griffin, L. D., Bell, I. M., & Thomas, G. M. H. (2022). Machine Learning of Raman Spectroscopy Data for Classifying Cancers: A Review of the Recent Literature. Diagnostics, 12(6), 1491. https://doi.org/10.3390/diagnostics12061491