
TP53 Expression and Mutational Analysis in Hematological Malignancy in Jeddah, Saudi Arabia


 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Cytogenetic and FISH Analysis
2.3. Next-Generation Sequencing Analysis
2.4. Data Analysis
2.5. Structural Analysis
3. Results
3.1. Clinical Characteristics of Patients
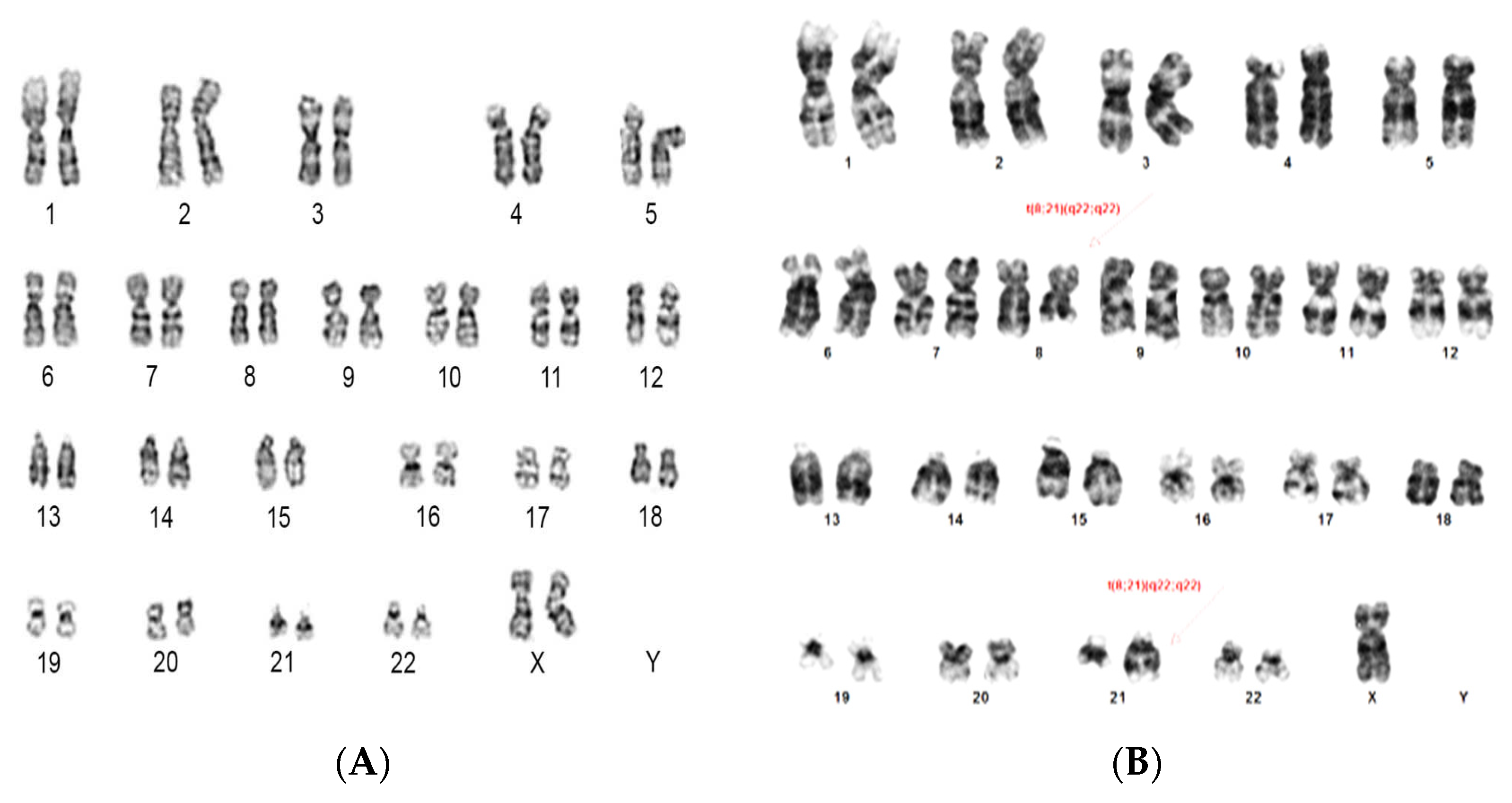
3.2. Cytogenetic and FISH Results
3.3. Next-Generation Sequencing Analysis
3.4. Correlation of TP53 Mutation with Cytogenetic & FISH Results
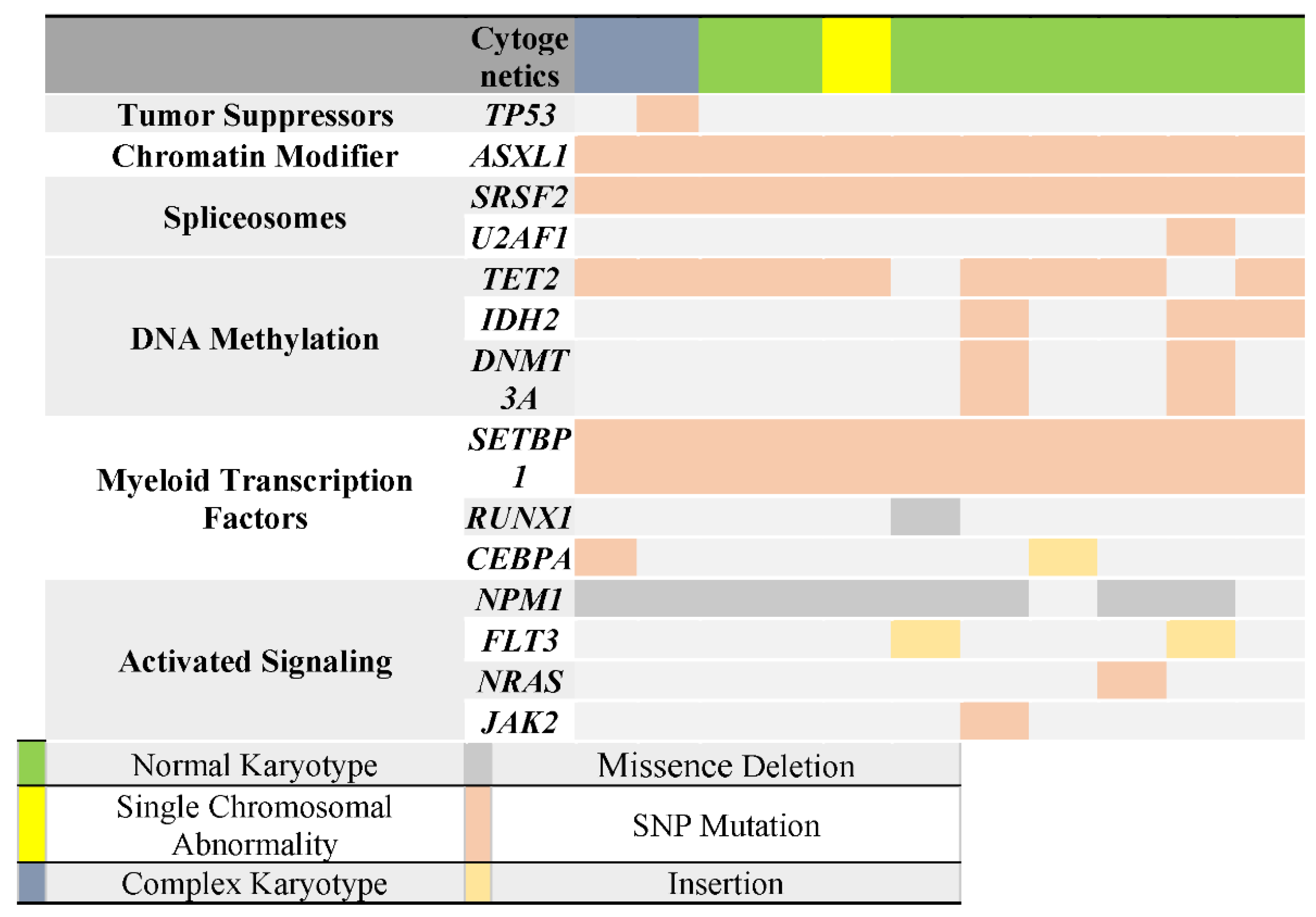
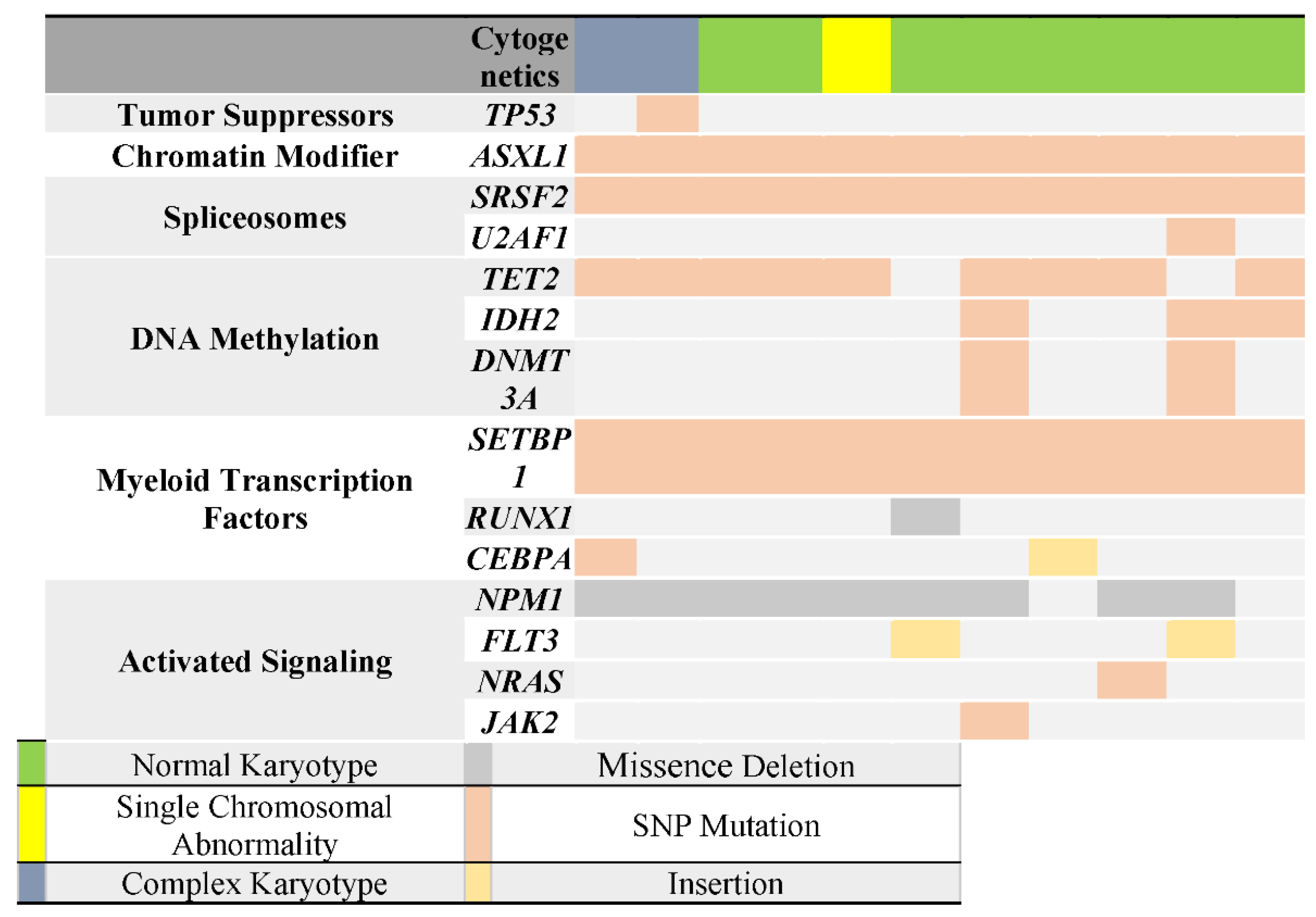
3.5. Correlation of TP53 Mutation with Other Genes Mutations
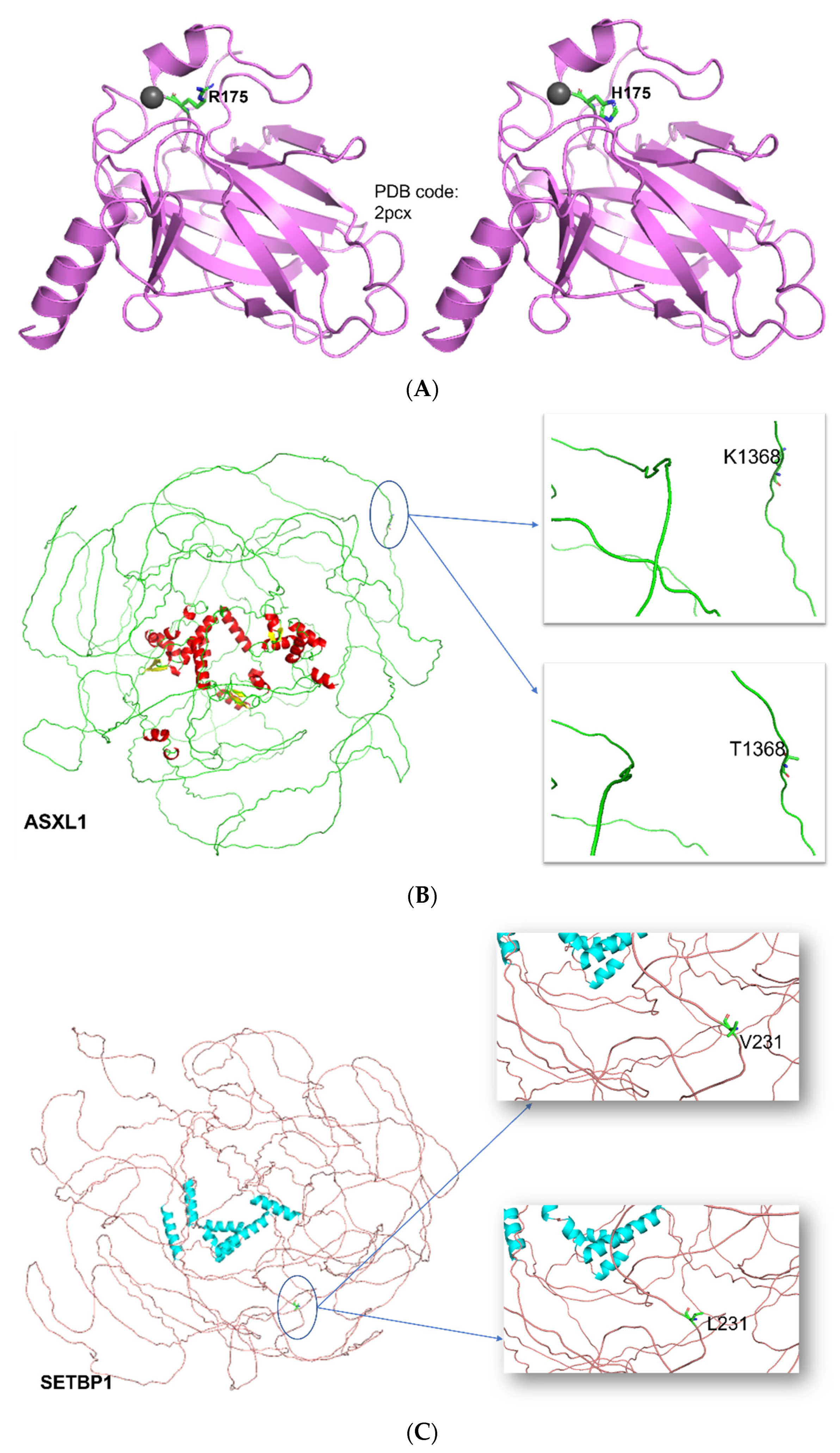
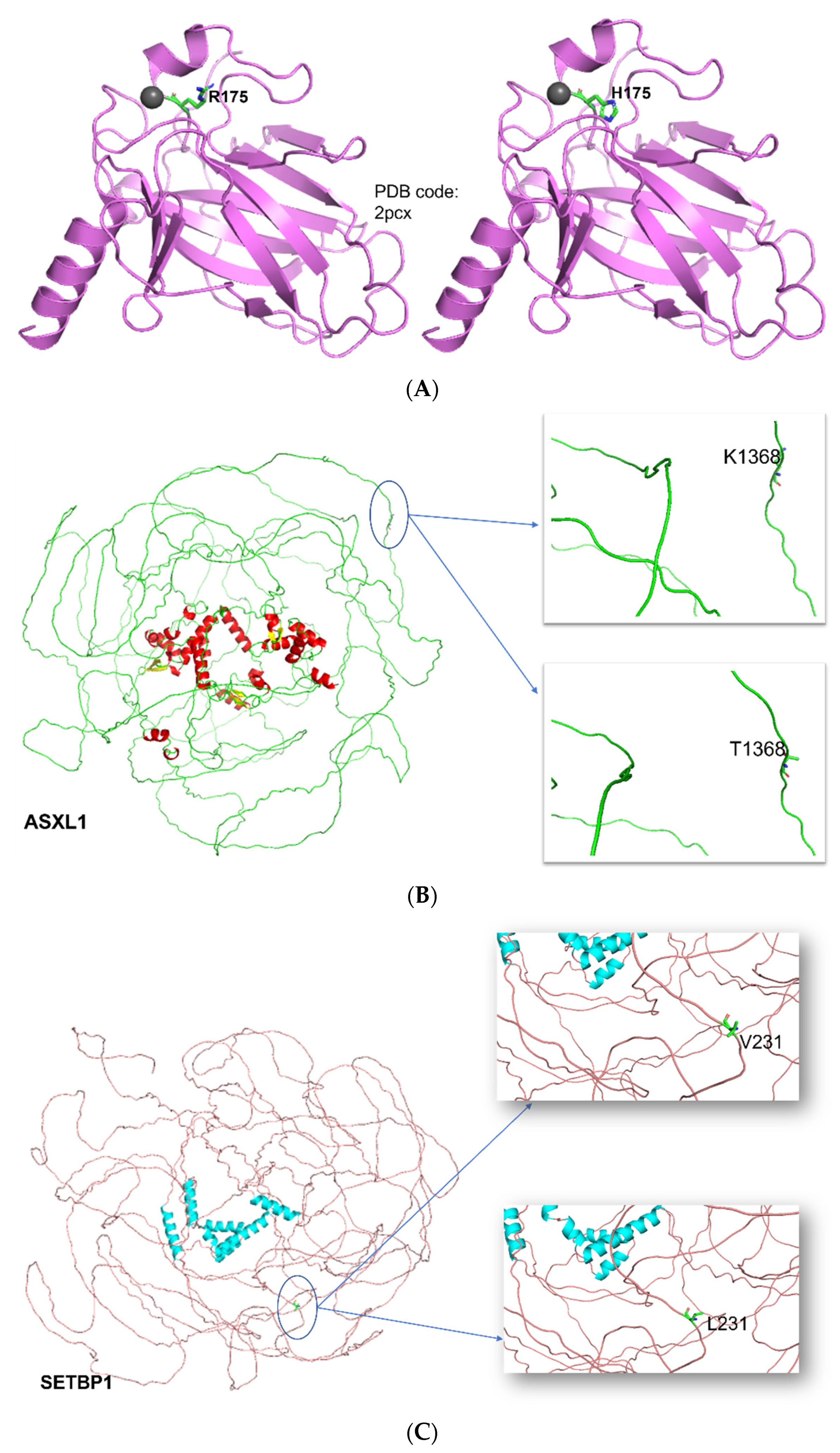
3.6. Structural and Functional Impact at the Protein Level
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preudhomme, C.; Fenaux, P. The Clinical Significance of Mutations of the p52 Tumour Suppressor Gene in Haematological Malignancies. Br. J. Haematol. 1997, 98, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Legros, Y.; Lubin, R.; Ory, K.; Schlichtholz, B. Multifactorial analysis of p53 alteration in human cancer: A review. Int. J. Cancer 1994, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wickremasinghe, R.; Prentice, A.; Steele, A. p53 and Notch signaling in chronic lymphocytic leukemia: Clues to identifying novel therapeutic strategies. Leukemia 2011, 25, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Fenaux, P.; Preudhomme, C.; Quiquandon, I.; Jonveaux, P.; Laï, J.L.; Vanrumbeke, M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. Mutations of the P53 gene in acute myeloid leukaemia. Br. J. Haematol. 1992, 80, 178–183. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Rotter, V.; Aloni-Grinstein, R.; Schwartz, D.; Elkind, N.; Simons, A.; Wolkowicz, R.; Lavigne, M.; Beserman, P.; Kapon, A.; Goldfinger, N. Does wild-type p53 play a role in normal cell differentiation? Semin. Cancer Biol. 1994, 5, 229–236. [Google Scholar]
- Wynford-Thomas, D. Cellular senescence and cancer. J. Pathol. 1999, 187, 100–111. [Google Scholar] [CrossRef]
- Agirre, X.; Novo, F.J.; Calasanz, M.J.; Larráyoz, M.J.; Lahortiga, I.; Valgañón, M.; García-Delgado, M.; Vizmanos, J.L. TP53 is frequently altered by methylation, mutation, and/or deletion in acute lymphoblastic leukaemia. Mol. Carcinog. 2003, 38, 201–208. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venot, C.; Maratrat, M.; Dureuil, C.; Conseiller, E.; Bracco, L.; Debussche, L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998, 17, 4668–4679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Chen, X. The C terminus of p53 family proteins is a cell fate determinant. Mol. Cell. Biol. 2005, 25, 2014–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.; Freeman, J.; Bovey, R.; Iggo, R. Regulation of specific DNA binding by p53: Evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene 1996, 12, 921–930. [Google Scholar]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.i.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation–acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [Green Version]
- Kruse, J.-P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Gaidano, G. Molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev. Hematol. 2012, 5, 593–602. [Google Scholar] [CrossRef]
- Hainaut, P.; Soussi, T.; Shomer, B.; Hollstein, M.; Greenblatt, M.; Hovig, E.; Harris, C.; Montesano, R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997, 25, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP 53 p. R273H and p. R273C but not p. R273G Enhance Cancer Cell Malignancy. Hum. Mutat. 2014, 35, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Schnittger, S.; Weissmann, S.; Kuznia, S.; Kern, W.; Kohlmann, A.; Haferlach, T.; Haferlach, C. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood J. Am. Soc. Hematol. 2014, 124, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, V.; Schnittger, S.; Kohlmann, A.; Eder, C.; Roller, A.; Dicker, F.; Schmid, C.; Wendtner, C.-M.; Staib, P.; Serve, H. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood J. Am. Soc. Hematol. 2012, 120, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood J. Am. Soc. Hematol. 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Cerri, M.; Deambrogi, C.; Sozzi, E.; Cresta, S.; Rasi, S.; De Paoli, L.; Spina, V.; Gattei, V.; Capello, D. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: Implications for overall survival and chemorefractoriness. Clin. Cancer Res. 2009, 15, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood J. Am. Soc. Hematol. 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Hof, J.; Krentz, S.; Van Schewick, C.; Körner, G.; Shalapour, S.; Rhein, P.; Karawajew, L.; Ludwig, W.-D.; Seeger, K.; Henze, G. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2011, 29, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Bartram, C.R.; Nakamura, H.; Hachiya, M.; Chen, D.-L.; Borenstein, J.; Miller, C.W.; Ludwig, L.; Hansen-Hagge, T.E.; Ludwig, W.-D. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood 1993, 82, 3163–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, C.; Schnittger, S.; Bursch, S.; Gerstner, D.; Hochhaus, A.; Berger, U.; Hehlmann, R.; Hiddemann, W.; Haferlach, T. Comparison of chromosome banding analysis, interphase-and hypermetaphase-FISH, qualitative and quantitative PCR for diagnosis and for follow-up in chronic myeloid leukemia: A study on 350 cases. Leukemia 2002, 16, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker, F.; Schnittger, S.; Haferlach, T.; Kern, W.; Schoch, C. Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: A study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood 2006, 108, 3152–3160. [Google Scholar] [CrossRef] [Green Version]
- Haferlach, C.; Bacher, U. Cytogenetic methods in chronic lymphocytic leukemia. In Cancer Cytogenetics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 119–130. [Google Scholar]
- Hastings, R.J.; Bown, N.; Tibiletti, M.G.; Debiec-Rychter, M.; Vanni, R.; Espinet, B.; Van Roy, N.; Roberts, P.; Van Den Berg-De-Ruiter, E.; Bernheim, A. Guidelines for cytogenetic investigations in tumours. Eur. J. Hum. Genet. 2016, 24, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, E.M.; Bethel, J.S.; McGlone, J.C. Introduction to Modern Photogrammetry; ACADEMIA: New York, NY, USA, 2001; p. 19. [Google Scholar]
- Suzuki, K.; Matsubara, H. Recent advances in p53 research and cancer treatment. J. Biomed. Biotechnol. 2011, 2011, 978312. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Peller, S.; Rotter, V. TP53 in hematological cancer: Low incidence of mutations with significant clinical relevance. Hum. Mutat. 2003, 21, 277–284. [Google Scholar] [CrossRef]
- Cazzola, A.; Schlegel, C.; Jansen, I.; Bochtler, T.; Jauch, A.; Krämer, A. TP53 deficiency permits chromosome abnormalities and karyotype heterogeneity in acute myeloid leukemia. Leukemia 2019, 33, 2619–2627. [Google Scholar] [CrossRef]
- Alwash, Y.; Khoury, J.D.; Tashakori, M.; Kanagal-Shamanna, R.; Daver, N.; Ravandi, F.; Kadia, T.M.; Konopleva, M.; Dinardo, C.D.; Issa, G.C. Development of TP53 Mutations Over the Course of Therapy for Acute Myeloid Leukemia. Am. J. Hematol. 2021, 96, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R. Applications of fluorescence in situ hybridization (FISH) in detecting genetic aberrations of medical significance. Biosci. Horiz. 2010, 3, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Moueden, A.; Benlaldj, D.; Boumeddane, A.; Seghier, F. Aberrant Expression of the p53 Tumor Suppressor Gene in Pediatric Acute Lymphoblastic Leukemia. J. Blood Lymph 2018, 8, 2. [Google Scholar] [CrossRef]
- Annooz, A.F.; Melconian, A.K.; Abdul-Majeed, B.A.; Jawad, A.M. Detection p53 gene deletion in hematological malignancies using fluorescence in situ hybridization: A pilot study. Pak. J. Biol. Sci. 2014, 17, 891–897. [Google Scholar] [CrossRef]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J. Clin. Oncol. 2018, 36, 591. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.J.; Choi, J.W.; Hong, M.H.; Jung, D.; Son, H.; Cho, E.K.; Min, Y.J.; Kim, S.-W.; Park, K.; Lee, S.S. Molecular characterization of lung adenocarcinoma from Korean patients using next generation sequencing. PLoS ONE 2019, 14, e0224379. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Misawa, S.; Horiike, S. TP53 mutations in myelodysplastic syndrome. Leuk. Lymphoma 1996, 23, 417–422. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gäken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B. TP 53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Mack, A.; Kucia, M.; Ratajczak, J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia 2018, 32, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- La Starza, R.; Matteucci, C.; Gorello, P.; Brandimarte, L.; Pierini, V.; Crescenzi, B.; Nofrini, V.; Rosati, R.; Gottardi, E.; Saglio, G. NPM1 deletion is associated with gross chromosomal rearrangements in leukemia. PLoS ONE 2010, 5, e12855. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Idossa, D.; Lasho, T.L.; Mudireddy, M.; Finke, C.; Shah, S.; Nicolosi, M.; Patnaik, M.M.; Pardanani, A.; Gangat, N. Mutations and karyotype in myelodysplastic syndromes: TP53 clusters with monosomal karyotype, RUNX1 with trisomy 21, and SF3B1 with inv (3)(q21q26. 2) and del (11q). Blood Cancer J. 2017, 7, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddel, H.K.; Bateman, E.D.; Becker, A.; Boulet, L.-P.; Cruz, A.A.; Drazen, J.M.; Haahtela, T.; Hurd, S.S.; Inoue, H.; De Jongste, J.C. A summary of the new GINA strategy: A roadmap to asthma control. Eur. Respir. J. 2015, 46, 622–639. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Kitaura, J.; Matsui, H.; Hou, H.A.; Chou, W.C.; Nagamachi, A.; Kawabata, K.C.; Togami, K.; Nagase, R.; Horikawa, S.; et al. SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia 2015, 29, 847–857. [Google Scholar] [CrossRef]
- Devillier, R.; Mas, V.M.-D.; Gelsi-Boyer, V.; Demur, C.; Murati, A.; Corre, J.; Prebet, T.; Bertoli, S.; Brecqueville, M.; Arnoulet, C.; et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget 2015, 6, 8388–8396. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Cassier, P.A.; Castets, M.; Belhabri, A.; Vey, N. Targeting apoptosis in acute myeloid leukaemia. Br. J. Cancer 2017, 117, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.F.; Nasrin, N.; Akhtar, M.; Hannan, M.A. Studies of the p53 gene mutation in Saudi non-Hodgkin’s lymphoma. Cancer Lett. 1996, 104, 225–231. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene List (Targeted Exons) | |||||||
|---|---|---|---|---|---|---|---|
| GENE | EXON | GENE | EXON | GENE | EXON | GENE | EXON |
| ASXL1 | 12 | EZH2 | 8, 17, 18 | MPL | 10 | SF3B1 | 13–15, 17 |
| CSF3R | 14, 17 | FLT3 | 14, 20 | NPM1 | 11 | SRSF2 | 1 |
| CBL | 8, 9 | IDH1 | 4 | NRAS | 2, 3 | TET2 | 3, 9, 10, 11 |
| FFCEBPA | 1 | IDH2 | 4 | RUNX1 | 3, 4, 8 | TP53 | 5–8 |
| DNMT3A | 4, 8, 13, 15, 16, 18–23 | JAK2 | 12, 14 | SETBP1 | 3 | U2AF1 | 2, 6 |
| Case N.O. | Age | Sex | Diagnosis | Cytogenetic Result |
|---|---|---|---|---|
| 1 | 2 | M | ALL | 46, XY |
| 2 | 2 | M | ALL | AL,57~43, XY, +X, dup (1) (q21q31), +4, +5, +6, +7, −8, +9, +10, +14, +17, +18, −19, −20, +21, +22 [cp50] |
| 3 | 5 | F | ALL | Leukemia,46, XX, der (19) t (1;19) (q25; p13.3) [17]/46, XX, idem, +der (21) t (1;21) (p13; p11.2) [14]/46, XX [19] |
| 4 | 5 | M | ALL | 46, XX [20] |
| 5 | 7 | F | ALL | 46, XX [20] |
| 6 | 9 | M | AML | 46, XY [20] |
| 7 | 12 | M | ALL | 46, XY, t (8; 21)(q22; q22) [29]/46, XY [21] |
| 8 | 31 | M | ALL | 46, XY [20] |
| 9 | 36 | F | AML | 46, XX [20] |
| 10 | 43 | M | AML | 46, XY [20] |
| 11 | 44 | F | ALL | ALL,45, XX, +X, −9, t (9;22) (q34; q11.2), −13[cp34]/45, XX, t (9;22) (q34; q11.2) [cp8]/46, XX[cp8] |
| 12 | 45 | F | ALL | 46, XX [20] |
| 13 | 59 | F | Lymphoma, NHL | 46, XX [20] |
| 14 | 63 | F | AML | 46, XX [20] |
| 15 | 65 | F | MDS | 65-58, XX, +1, +2, der(2) t(2;5) (q12; q37), +5, +6, +8, +9, +10, +11, der(17) t (12;17) (p10; p10), +13, del (13) (q21), +21, +21 [cp50] |
| 16 | 72 | M | AML | 45, XY,der (7;12), (q11.2; p12) [30] |
| 17 | 13 Y | F | AML | 46, XX [20] |
| 18 | 35 Y | F | AML | 46, XY [20] |
| 19 | 42 Y | M | AML | 46, XX [20] |
| 20 | 69 Y | M | AML | 46, XY [20] |
| Diagnosis | Cytogenetic | FISH |
|---|---|---|
| NHL (1), ALL (5), AML (8) | Normal karyotype (14) | NHL TP53 deletion (1/1) (11%) |
| ALL TP53 deletion (2/5) (20–22%) | ||
| AML TP53 deletion (1/8) (11%) | ||
| AML TP53 trisomy (1/8) (20%) | ||
| ALL (1), AML (1) | Single abnormality (2) | ALL TP53 deletion (1/1) (22%) |
| AML TP53 deletion (0/1) | ||
| MDS (1), ALL (3) | Complex karyotype (4) | MDS (1/1) TP53 trisomy (30%) |
| ALL (2/3) TP53 deletion (45%) |
| Impacted Gene | TP53 |
|---|---|
| Type of Mutation | Missense mutation (Heterozygous) |
| Chromosome | 17 |
| Ref. Allele | T |
| Alt. Allele | C |
| Function Class | Missense |
| AA | H175R |
| Codon | cAt/cGt |
| Quality | Pass |
| Allele Frequency | 0.447 |
| Number of Variant Alleles | 10,232 |
| Filtered Read Depth (per sample) | 22,870 |
| Effect | UNKNOWN |
| Exon ID | NM_001126118.ex.5 |
| Gene | Total No. of Mutation (n = 10) | TP53-Mutated (n = 1) | TP53-wt (n = 9) |
|---|---|---|---|
| ASXL1 | 10 | 1 | 9 |
| CEBPA | 2 | 0 | 2 |
| DNMT3A | 2 | 0 | 2 |
| FLT3 | 2 | 0 | 2 |
| IDH2 | 3 | 0 | 3 |
| JAK2 | 1 | 0 | 1 |
| NPM1 | 8 | 1 | 7 |
| RUNX1 | 4 | 0 | 4 |
| SETBP1 | 10 | 1 | 9 |
| SRSF2 | 10 | 1 | 9 |
| TET2 | 8 | 1 | 7 |
| U2AF1 | 1 | 0 | 1 |
| NRAS | 1 | 0 | 1 |
| Sample No. | Age | Sex | Diagnosis | Gene | Type of Mutation |
|---|---|---|---|---|---|
| 1 | 5 Y | F | ALL | ASXL1 | Missense (L815P) |
| SETBP1 | Silent (S1275) | ||||
| 2 | 65 Y | F | MDS | ASXL1 | Missense (L815P) Missense (K1368T) |
| SETBP1 | Missense (V231L) Silent (S1275) | ||||
| 3 | 59 Y | F | Lymphoma, NHL | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Missense (V1101I) Silent (S1275) | ||||
| 4 | 63 Y | F | AML | ASXL1 | Missense (L815P) |
| SETBP1 | Silent (S1275) | ||||
| 5 | 36 Y | F | AML | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Silent (H1206) Silent (S1275) Silent (L1278) | ||||
| 6 | 43 Y | M | AML | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Silent (S1275) | ||||
| 7 | 13 Y | F | AML | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Silent (H1206) Silent (S1275) | ||||
| 8 | 35 Y | F | AML | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Missense (V1101I) Silent (S1275) | ||||
| 9 | 42 Y | M | AML | ASXL1 | Missense (L815P) Silent (S1253) |
| SETBP1 | Silent (S1275) | ||||
| 10 | 69 Y | M | AML | ASXL1 | Missense (L815P) |
| SETBP1 | Silent (S1275) |
| Gene-Mutation | Polyphen-2 | I-Mutant 2.0 | ClinVar |
|---|---|---|---|
| TP53-R175H | POSSIBLY DAMAGING score: 0.881 (sensitivity: 0.82; specificity: 0.94) | Decrease in stability ΔΔG = −1.35 Kcal/mol | PATHOGENIC |
| ASXL1-K1368T | BENIGN score: 0.091 (sensitivity: 0.93; specificity: 0.85) | Decrease in stability ΔΔG = −0.63 Kcal/mol | - |
| SETBP1-V231L | BENIGN score: 0.006 (sensitivity: 0.97; specificity: 0.75) | Decrease in stability ΔΔG = −0.19 Kcal/mol | BENIGN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhatabi, H.; Yasin, E.B.; Mirza, Z.; Alserihi, R.; Felimban, R.; Elaimi, A.; Shaabad, M.; Alharbi, L.; Ahmed, H.; Alameer, A.M.; et al. TP53 Expression and Mutational Analysis in Hematological Malignancy in Jeddah, Saudi Arabia. Diagnostics 2022, 12, 724. https://doi.org/10.3390/diagnostics12030724
Alkhatabi H, Yasin EB, Mirza Z, Alserihi R, Felimban R, Elaimi A, Shaabad M, Alharbi L, Ahmed H, Alameer AM, et al. TP53 Expression and Mutational Analysis in Hematological Malignancy in Jeddah, Saudi Arabia. Diagnostics. 2022; 12(3):724. https://doi.org/10.3390/diagnostics12030724
Chicago/Turabian StyleAlkhatabi, Heba, Elrashed B. Yasin, Zeenat Mirza, Raed Alserihi, Raed Felimban, Aisha Elaimi, Manal Shaabad, Lina Alharbi, Hameeda Ahmed, Abdulrahman M. Alameer, and et al. 2022. "TP53 Expression and Mutational Analysis in Hematological Malignancy in Jeddah, Saudi Arabia" Diagnostics 12, no. 3: 724. https://doi.org/10.3390/diagnostics12030724
APA StyleAlkhatabi, H., Yasin, E. B., Mirza, Z., Alserihi, R., Felimban, R., Elaimi, A., Shaabad, M., Alharbi, L., Ahmed, H., Alameer, A. M., Mathkoor, A. E., & Barefah, A. S. (2022). TP53 Expression and Mutational Analysis in Hematological Malignancy in Jeddah, Saudi Arabia. Diagnostics, 12(3), 724. https://doi.org/10.3390/diagnostics12030724