
NGS Analysis Revealed Digenic Heterozygous GCK and HNF1A Variants in a Child with Mild Hyperglycemia: A Case Report


 ,
, 
Abstract
1. Introduction
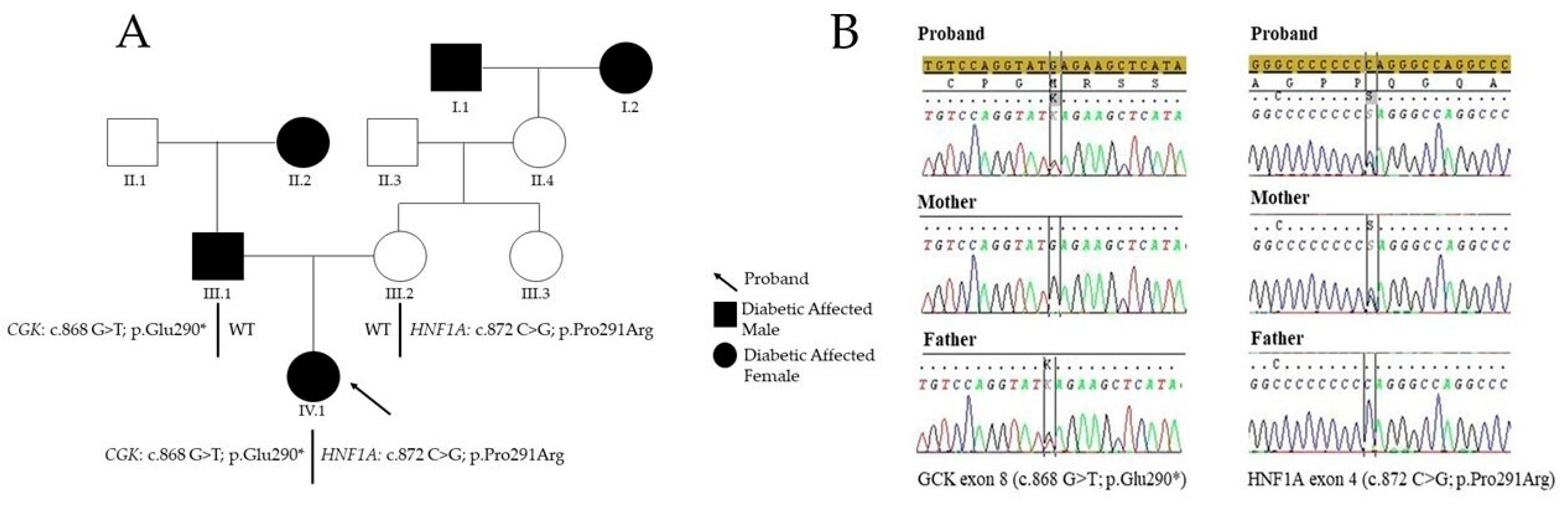
2. Case Presentation
3. Diagnostic Assessment
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mozzillo, E.; Salzano, G.; Barbetti, F.; Maffeis, C.; Lombardo, F.; Franzese, A.; Delvecchio, M.; Marigliano, M. Survey on Etiological Diagnosis of Diabetes in 1244 Italian Diabetic Children and Adolescents: Impact of Access to Genetic Testing. Diabetes Res. Clin. Pract. 2015, 107, e15–e18. [Google Scholar] [CrossRef]
- Vaxillaire, M.; Bonnefond, A.; Liatis, S.; Ben Salem Hachmi, L.; Jotic, A.; Boissel, M.; Gaget, S.; Durand, E.; Vaillant, E.; Derhourhi, M.; et al. Monogenic Diabetes Characteristics in a Transnational Multicenter Study from Mediterranean Countries. Diabetes Res. Clin. Pract. 2021, 171, 108553. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I.; Polonsky, K.S. Molecular Mechanisms and Clinical Pathophysiology of Maturity-Onset Diabetes of the Young. N. Engl. J. Med. 2001, 345, 971–980. [Google Scholar] [CrossRef]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The Epidemiology, Molecular Pathogenesis, Diagnosis, and Treatment of Maturity-Onset Diabetes of the Young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T. European Molecular Genetics Quality Network (EMQN) MODY group Best Practice Guidelines for the Molecular Genetic Diagnosis of Maturity-Onset Diabetes of the Young. Diabetologia 2008, 51, 546–553. [Google Scholar] [CrossRef]
- Capuano, M.; Garcia-Herrero, C.M.; Tinto, N.; Carluccio, C.; Capobianco, V.; Coto, I.; Cola, A.; Iafusco, D.; Franzese, A.; Zagari, A.; et al. Glucokinase (GCK) Mutations and Their Characterization in MODY2 Children of Southern Italy. PLoS ONE 2012, 7, e38906. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Mozzillo, E.; Salzano, G.; Iafusco, D.; Frontino, G.; Patera, P.I.; Rabbone, I.; Cherubini, V.; Grasso, V.; Tinto, N.; et al. Monogenic Diabetes Accounts for 6.3% of Cases Referred to 15 Italian Pediatric Diabetes Centers During 2007 to 2012. J. Clin. Endocrinol. Metab. 2017, 102, 1826–1834. [Google Scholar] [CrossRef]
- Estalella, I.; Rica, I.; Perez de Nanclares, G.; Bilbao, J.R.; Vazquez, J.A.; San Pedro, J.I.; Busturia, M.A.; Castaño, L. Spanish MODY Group Mutations in GCK and HNF-1alpha Explain the Majority of Cases with Clinical Diagnosis of MODY in Spain. Clin. Endocrinol. 2007, 67, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Schober, E.; Rami, B.; Grabert, M.; Thon, A.; Kapellen, T.; Reinehr, T.; Holl, R.W. DPV-Wiss Initiative of the German Working Group for Paediatric Diabetology and Phenotypical Aspects of Maturity-Onset Diabetes of the Young (MODY Diabetes) in Comparison with Type 2 Diabetes Mellitus (T2DM) in Children and Adolescents: Experience from a Large Multicentre Database. Diabet. Med. 2009, 26, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Carmody, D.; Naylor, R.N.; Bell, C.D.; Berry, S.; Montgomery, J.T.; Tadie, E.C.; Hwang, J.L.; Greeley, S.A.W.; Philipson, L.H. GCK-MODY in the US National Monogenic Diabetes Registry: Frequently Misdiagnosed and Unnecessarily Treated. Acta Diabetol. 2016, 53, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Tinto, N.; Zagari, A.; Capuano, M.; De Simone, A.; Capobianco, V.; Daniele, G.; Giugliano, M.; Spadaro, R.; Franzese, A.; Sacchetti, L. Glucokinase Gene Mutations: Structural and Genotype-Phenotype Analyses in MODY Children from South Italy. PLoS ONE 2008, 3, e1870. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, O.; Tinto, N.; Franzese, A.; Iafusco, F.; Festa, C.; Mozzillo, E.; Napoli, A.; Iafusco, D. Glucokinase Deficit and Birthweight: Does Maternal Hyperglycemia Always Meet Fetal Needs? Acta Diabetol. 2018, 55, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Salzano, G.; Bonura, C.; Cauvin, V.; Cherubini, V.; d’Annunzio, G.; Franzese, A.; Giglio, S.; Grasso, V.; Graziani, V.; et al. Can HbA1c Combined with Fasting Plasma Glucose Help to Assess Priority for GCK-MODY vs HNF1A-MODY Genetic Testing? Acta Diabetol. 2018, 55, 981–983. [Google Scholar] [CrossRef]
- Iafusco, F.; De Sanctis, P.; Pirozzi, D.; Capone, S.; Lombardo, B.; Gambale, A.; Confetto, S.; Zanfardino, A.; Iolascon, A.; Pastore, L.; et al. Molecular Diagnosis of MODY3 Permitted to Reveal a de Novo 12q24.31 Deletion and to Explain a Complex Phenotype in a Young Diabetic Patient. Clin. Chem. Lab. Med. 2019, 57, e306–e310. [Google Scholar] [CrossRef]
- Pinelli, M.; Acquaviva, F.; Barbetti, F.; Caredda, E.; Cocozza, S.; Delvecchio, M.; Mozzillo, E.; Pirozzi, D.; Prisco, F.; Rabbone, I.; et al. Identification of Candidate Children for Maturity-Onset Diabetes of the Young Type 2 (MODY2) Gene Testing: A Seven-Item Clinical Flowchart (7-IF). PLoS ONE 2013, 8, e79933. [Google Scholar] [CrossRef]
- Szopa, M.; Matejko, B.; Ucieklak, D.; Uchman, A.; Hohendorff, J.; Mrozińska, S.; Głodzik, W.; Zapała, B.; Płatek, T.; Solecka, I.; et al. Quality of Life Assessment in Patients with HNF1A-MODY and GCK-MODY. Endocrine 2019, 64, 246–253. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and Clinical Significance of Genetic Screening in Elite and Amateur Athletes. Eur. J. Prev. Cardiol. 2020, 2047487320934265. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.L.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence Variant Classification and Reporting: Recommendations for Improving the Interpretation of Cancer Susceptibility Genetic Test Results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Iafusco, D.; Liguori, R.; Ferrigno, M.; Galderisi, A.; Vitale, D.; Simonelli, F.; Landolfo, P.; Prisco, F.; Masullo, M.; et al. Mitochondrial Diabetes in Children: Seek and You Will Find It. PLoS ONE 2012, 7, e34956. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.T.; Vasta, V.; Zhang, M.; Narayanan, J.; Gerrits, P.; Hahn, S.H. Molecular Genetic Testing of Patients with Monogenic Diabetes and Hyperinsulinism. Mol. Genet. Metab. 2015, 114, 451–458. [Google Scholar] [CrossRef]
- Kim, S.-H.; Ma, X.; Klupa, T.; Powers, C.; Pezzolesi, M.; Warram, J.H.; Rich, S.S.; Krolewski, A.S.; Doria, A. Genetic Modifiers of the Age at Diagnosis of Diabetes (MODY3) in Carriers of Hepatocyte Nuclear Factor-1alpha Mutations Map to Chromosomes 5p15, 9q22, and 14q24. Diabetes 2003, 52, 2182–2186. [Google Scholar] [CrossRef] [PubMed]
- Miedzybrodzka, Z.; Hattersley, A.T.; Ellard, S.; Pearson, D.; de Silva, D.; Harvey, R.; Haites, N. Non-Penetrance in a MODY 3 Family with a Mutation in the Hepatic Nuclear Factor 1alpha Gene: Implications for Predictive Testing. Eur. J. Hum. Genet. 1999, 7, 729–732. [Google Scholar] [CrossRef][Green Version]
- Locke, J.M.; Saint-Martin, C.; Laver, T.W.; Patel, K.A.; Wood, A.R.; Sharp, S.A.; Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T.; Harries, L.W.; et al. The Common HNF1A Variant I27L Is a Modifier of Age at Diabetes Diagnosis in Individuals with HNF1A-MODY. Diabetes 2018, 67, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Spiro, A.J.; Vu, K.N.; Warnock, A.L. An Atypical HNF4A Mutation Which Does Not Conform to the Classic Presentation of HNF4A-MODY. Case Rep. Endocrinol. 2018, 2018, 1560472. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.W.; Colclough, K.; Shepherd, M.; Patel, K.; Houghton, J.A.L.; Dusatkova, P.; Pruhova, S.; Morris, A.D.; Palmer, C.N.; McCarthy, M.I.; et al. The Common p.R114W HNF4A Mutation Causes a Distinct Clinical Subtype of Monogenic Diabetes. Diabetes 2016, 65, 3212–3217. [Google Scholar] [CrossRef]
- Flannick, J.; Beer, N.L.; Bick, A.G.; Agarwala, V.; Molnes, J.; Gupta, N.; Burtt, N.P.; Florez, J.C.; Meigs, J.B.; Taylor, H.; et al. Assessing the Phenotypic Effects in the General Population of Rare Variants in Genes for a Dominant Mendelian Form of Diabetes. Nat. Genet. 2013, 45, 1380–1385. [Google Scholar] [CrossRef]
- Najmi, L.A.; Aukrust, I.; Flannick, J.; Molnes, J.; Burtt, N.; Molven, A.; Groop, L.; Altshuler, D.; Johansson, S.; Bjørkhaug, L.; et al. Functional Investigations of HNF1A Identify Rare Variants as Risk Factors for Type 2 Diabetes in the General Population. Diabetes 2017, 66, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.K.; Ellard, S.; Standiford, D.; Pihoker, C.; Gilliam, L.K.; Hattersley, A.; Dolan, L.M. Digenic Heterozygous HNF1A and HNF4A Mutations in Two Siblings with Childhood-Onset Diabetes. Pediatr. Diabetes 2013, 14, 535–538. [Google Scholar] [CrossRef]
- Karges, B.; Bergmann, C.; Scholl, K.; Heinze, E.; Rasche, F.M.; Zerres, K.; Debatin, K.-M.; Wabitsch, M.; Karges, W. Digenic Inheritance of Hepatocyte Nuclear Factor-1alpha and -1beta with Maturity-Onset Diabetes of the Young, Polycystic Thyroid, and Urogenital Malformations. Diabetes Care 2007, 30, 1613–1614. [Google Scholar] [CrossRef] [PubMed]
- Forlani, G.; Zucchini, S.; Di Rocco, A.; Di Luzio, R.; Scipione, M.; Marasco, E.; Romeo, G.; Marchesini, G.; Mantovani, V. Double Heterozygous Mutations Involving Both HNF1A/MODY3 and HNF4A/MODY1 Genes: A Case Report. Diabetes Care 2010, 33, 2336–2338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- López-Garrido, M.P.; Herranz-Antolín, S.; Alija-Merillas, M.J.; Giralt, P.; Escribano, J. Co-Inheritance of HNF1a and GCK Mutations in a Family with Maturity-Onset Diabetes of the Young (MODY): Implications for Genetic Testing. Clin. Endocrinol. 2013, 79, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.-C.; Liu, Y.-Y.; Li, X.; Wang, W.-X.; Irwin, D.M.; Zhang, Y.-P. Identification of HNF4A Mutation p.T130I and HNF1A Mutations p.I27L and p.S487N in a Han Chinese Family with Early-Onset Maternally Inherited Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 3582616. [Google Scholar] [CrossRef]


{kind=link}
| NGS GENE PANEL | ||||||
|---|---|---|---|---|---|---|
| ABCC8 | CEL | GCK | HNF4A | KLF11 | PDX1 | SLC16A1 |
| AKT2 | CISD2 | GLIS3 | IER3IP1 | LMNA | PLIN1 | SLC19A2 |
| APPL1 | EIF2AK3 | GLUD1 | INS | MAFA | POLD1 | SLC9B1 |
| ASB14 | FOXP3 | HADH | INS-IGF2 | NEUROD1 | PPARG | TRMT10A |
| BLK | GATA4 | HNF1A | INSR | NEUROG3 | PTF1A | WFS1 |
| C12orf43 | GATA6 | HNF1B | KCNJ11 | PAX4 | RFX6 | ZFP57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iafusco, F.; Maione, G.; Mazzaccara, C.; Di Candia, F.; Mozzillo, E.; Franzese, A.; Tinto, N. NGS Analysis Revealed Digenic Heterozygous GCK and HNF1A Variants in a Child with Mild Hyperglycemia: A Case Report. Diagnostics 2021, 11, 1164. https://doi.org/10.3390/diagnostics11071164
Iafusco F, Maione G, Mazzaccara C, Di Candia F, Mozzillo E, Franzese A, Tinto N. NGS Analysis Revealed Digenic Heterozygous GCK and HNF1A Variants in a Child with Mild Hyperglycemia: A Case Report. Diagnostics. 2021; 11(7):1164. https://doi.org/10.3390/diagnostics11071164
Chicago/Turabian StyleIafusco, Fernanda, Giovanna Maione, Cristina Mazzaccara, Francesca Di Candia, Enza Mozzillo, Adriana Franzese, and Nadia Tinto. 2021. "NGS Analysis Revealed Digenic Heterozygous GCK and HNF1A Variants in a Child with Mild Hyperglycemia: A Case Report" Diagnostics 11, no. 7: 1164. https://doi.org/10.3390/diagnostics11071164
APA StyleIafusco, F., Maione, G., Mazzaccara, C., Di Candia, F., Mozzillo, E., Franzese, A., & Tinto, N. (2021). NGS Analysis Revealed Digenic Heterozygous GCK and HNF1A Variants in a Child with Mild Hyperglycemia: A Case Report. Diagnostics, 11(7), 1164. https://doi.org/10.3390/diagnostics11071164