
Uranium Extraction from Salt Water Using Formo-Phenolic Resin Containing Amido-β-phosphonic Acid Chelating Moiety


 and
and
Abstract

1. Introduction
2. Experimental Part
2.1. Materials and Methods
2.2. Preparation of Amido-β-phosphonic Monomers
2.3. General Procedure for the Synthesis of Chelating Resin
2.4. Batch Experiments
- (i)
- Solution 1: a solution simulating seawater doped with 190 mg/L of uranium (8.0 × 10−4 mol/L), 259 mg/L of carbonates (4.3 × 10−3 M), and 2000 mg/L of sodium (8.7 × 10−2 M) at pH = 8.2 ± 0.1;
- (ii)
- Solution 2: a solution simulating seawater doped with 1 to 800 mg/L of uranium with a molar ratio fixed at around [Na]/[U] = 120 at pH = 8.2 ± 0.1;
- (iii)
- Solution 3: a solution simulating seawater with salinity at around 36 g/L (24.53 g NaCl, MgCl2 5.20 g/L, Na2SO4 4.09 g/L, CaCl2 1.16 g/L, KCl 0.695 g/L, NaHCO3 0.201 g/L, KBr 0.101 g/L) doped with 95 mg/L of uranium (4.0 × 10−4 mol/L), 96 mg/L of copper (1.5 × 10−3 mol/L), 97 mg/L of zinc (1.5 × 10−3 mol/L) and 39 mg/L of vanadium (7.7 × 10−4 mol/L) at pH = 5.9 ± 0.1;
- (iv)
- Solution 4: a solution simulating seawater with salinity at around 36 g/L (24.53 g NaCl, MgCl2 5.20 g/L, Na2SO4 4.09 g/L, CaCl2 1.16 g/L, KCl 0.695 g/L, NaHCO3 0.201 g/L, KBr 0.101 g/L) doped with 105 mg/L of uranium (4.4 × 10−4 mol/L) at pH = 8.2 ± 0.1.
- -
- The cation uptake capacity Qads (mg/g) was calculated using the equation:
- -
- , where Ci is the initial concentration of the metal ion in solution, Cf is the residual metal ion concentration, and V/m is the ratio of the solution volume by the resin’s mass;
- -
- The adsorption efficiency E (%) was calculated according to the following equation: ;
- -
- The distribution coefficient, denoted KD and expressed in mL/g, represents the ratio between the quantity of cation uptake by the resin and the quantity of cation remaining in solution after extraction, and was determined by the following formula:;
- -
- The separation factor FSU/M, with M the metal competitor, represents the ratio between the KD of uranium and the KD of the other metal. FSU/M allows for quantification of the selectivity of a resin to extract uranium rather than other metals and was calculated by the following formula: ;
- -
- The back extraction or stripping efficiency S (%) is defined by the following equation: , where Qe is the concentration of the metal ion loaded into the polymer at equilibrium and Qf is the residual metal ion concentration in the polymer after the release.
2.5. Measure of Amido-β-phosphonic Ratio in Resin
3. Results and Discussion
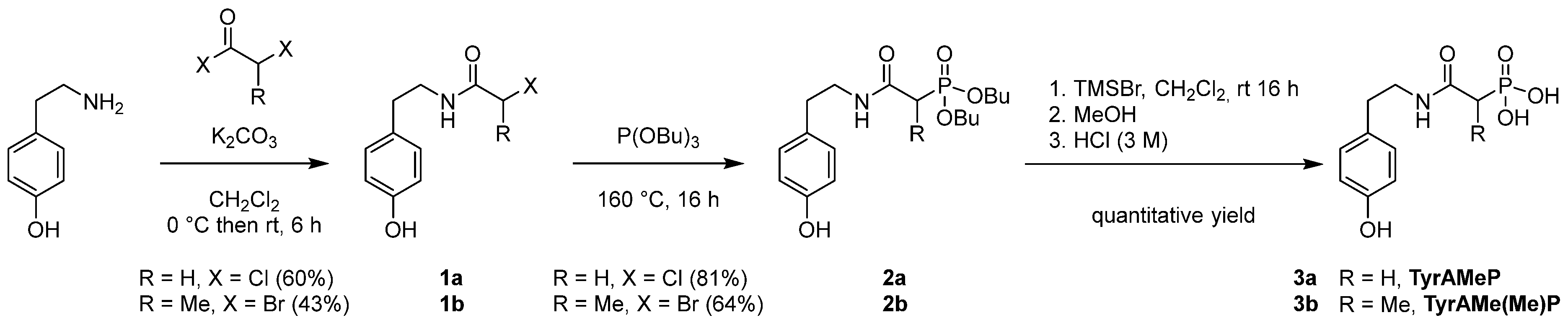
3.1. Synthesis of Amido-β-phosphonic Monomers
3.2. Chelating Thermosetting Resin Preparation
3.3. Characterization of Thermosetting Resins
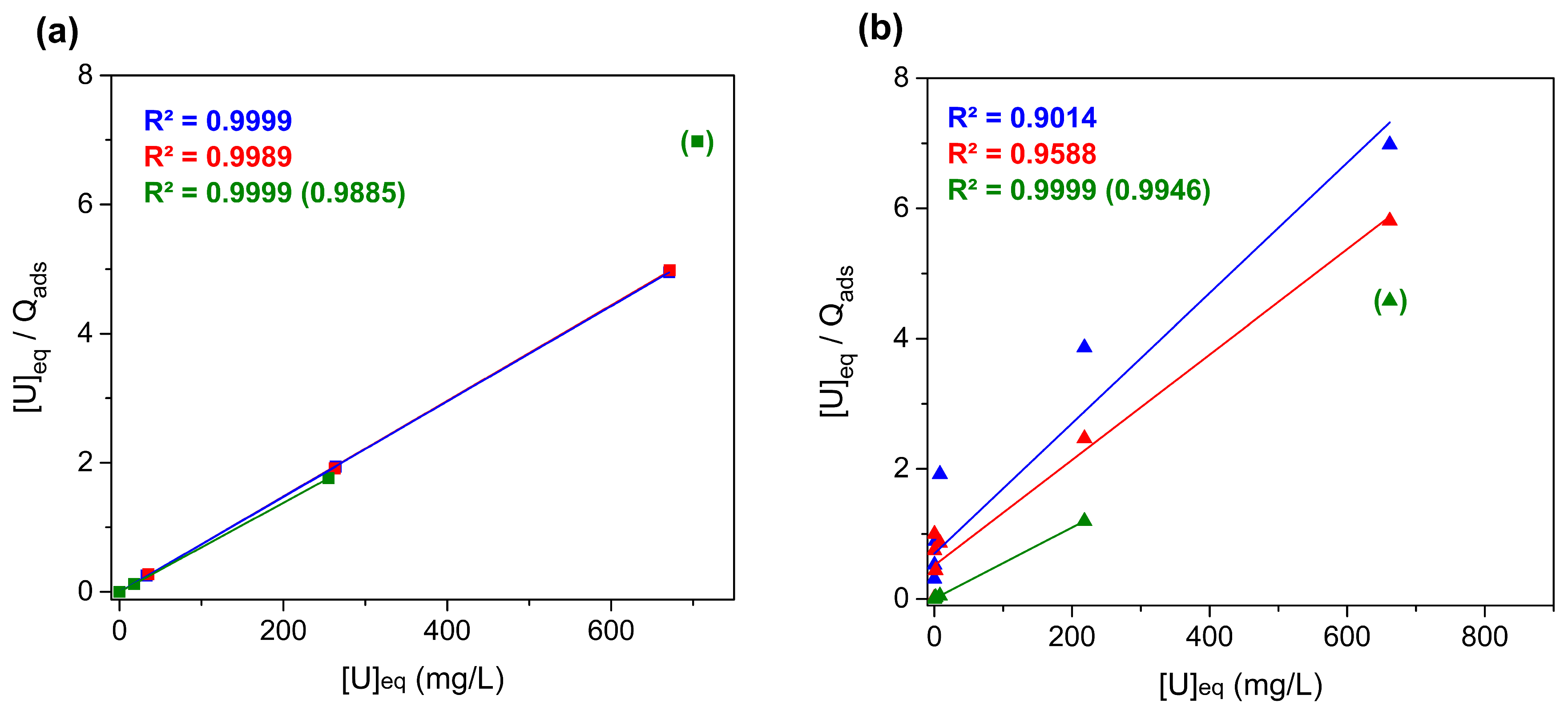
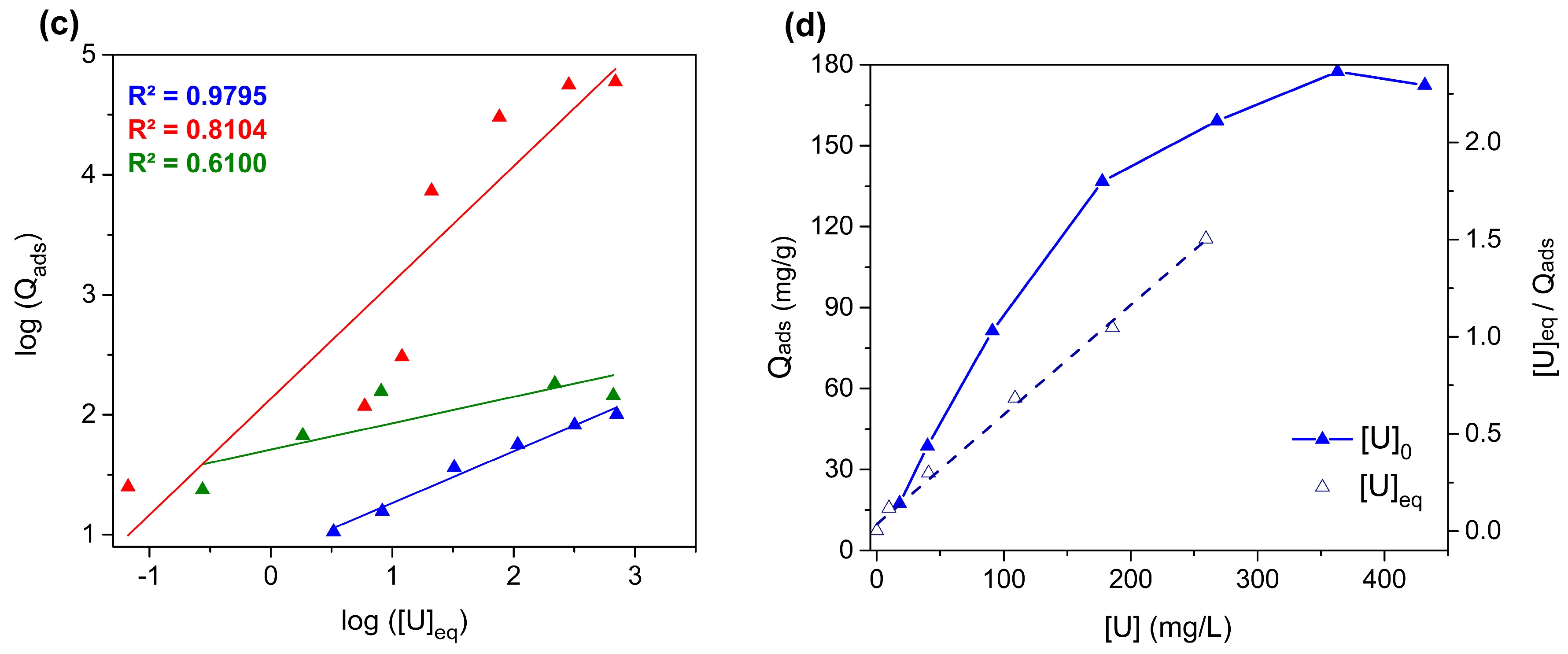
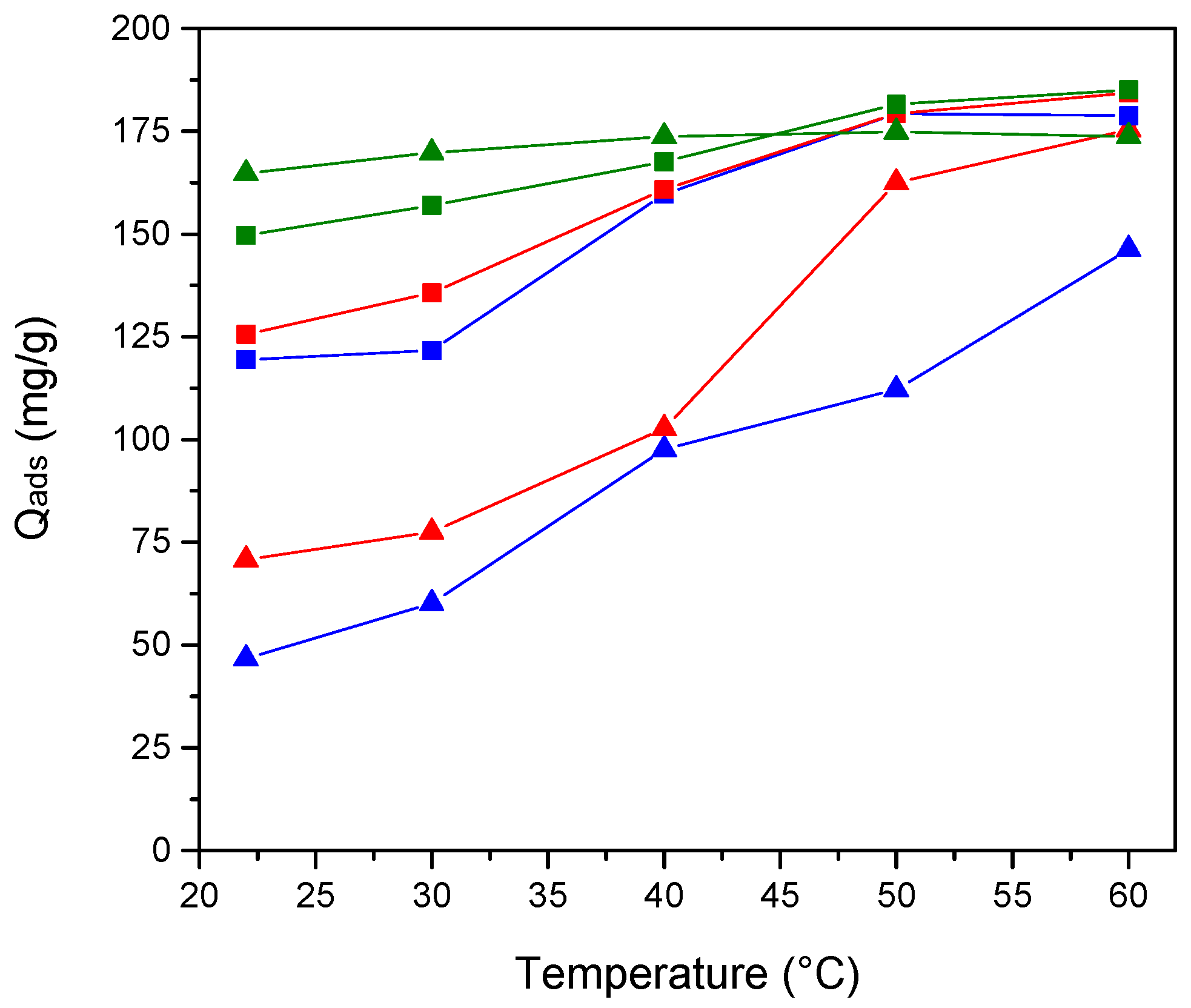
3.4. Studies of Uranium Extraction Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD. Uranium 2020: Resources, Production and Demand; NEA No. 7551; OECD Publishing: Paris, France, 2020. [Google Scholar]
- Wilson, J.D.; Webster, R.K.; Milner, G.W.C.; Barnett, G.A.; Smales, A.A. A comparison of three methods of determining the concentration of uranium in sea water. Anal. Chim. Acta 1960, 23, 505–514. [Google Scholar] [CrossRef]
- Sodaye, H.; Nisan, S.; Poletiko, C.; Prabhakar, S.; Tewari, P.K. Extraction of uranium from the concentrated brine rejected by integrated nuclear desalination plants. Desalination 2009, 235, 9–32. [Google Scholar] [CrossRef]
- Kim, J.; Tsouris, C.; Mayes, R.T.; Oyola, Y.; Saito, T.; Janke, C.J.; Dai, S.; Schneider, E.; Sachde, D. Recovery of Uranium from Seawater: A Review of Current Status and Future Research Needs. Sep. Sci. Technol. 2013, 48, 367–387. [Google Scholar] [CrossRef]
- Xing, C.; Bernicot, B.; Arrachart, G.; Pellet-Rostaing, S. Application of ultra/nano filtration membrane in uranium rejection from fresh and salt waters. Sep. Purif. Technol. 2023, 314, 123543. [Google Scholar] [CrossRef]
- Komjarova, I.; Blust, R. Comparison of liquid–liquid extraction, solid-phase extraction and co-precipitation preconcentration methods for the determination of cadmium, copper, nickel, lead and zinc in seawater. Anal. Chim. Acta 2006, 576, 221–228. [Google Scholar] [CrossRef]
- Cinnéide, S.; Scanlan, J.; Hynes, M. Equilibria in uranyl carbonate systems—I: The overall stability constant of UO2(CO3)34−. J. Inorg. Nucl. Chem. 1975, 37, 1013–1018. [Google Scholar] [CrossRef]
- Saito, K.; Miyauchi, T. Chemical Forms of Uranium in Artificial Seawater. J. Nucl. Sci. Technol. 1982, 19, 145–150. [Google Scholar] [CrossRef]
- Endrizzi, F.; Rao, L. Chemical Speciation of Uranium(VI) in Marine Environments: Complexation of Calcium and Magnesium Ions with [(UO2)(CO3)3]4− and the Effect on the Extraction of Uranium from Seawater. Chem.—Eur. J. 2014, 20, 14499–14506. [Google Scholar] [CrossRef]
- Schenk, H.J.; Astheimer, L.; Witte, E.G.; Schwochau, K. Development of Sorbers for the Recovery of Uranium from Seawater. 1. Assessment of Key Parameters and Screening Studies of Sorber Materials. Sep. Sci. Technol. 1982, 17, 1293–1308. [Google Scholar] [CrossRef]
- Kabay, N.; Egawa, H. Chelating Polymers for Recovery of Uranium from Seawater. Sep. Sci. Technol. 1994, 29, 135–150. [Google Scholar] [CrossRef]
- Seko, N.; Katakai, A.; Hasegawa, S.; Tamada, M.; Kasai, N.; Takeda, H.; Sugo, T.; Saito, K. Aquaculture of Uranium in Seawater by a Fabric-Adsorbent Submerged System. Nucl. Technol. 2003, 144, 274–278. [Google Scholar] [CrossRef]
- Zhang, A.; Uchiyama, G.; Asakura, T. pH Effect on the uranium adsorption from seawater by a macroporous fibrous polymeric material containing amidoxime chelating functional group. React. Funct. Polym. 2005, 63, 143–153. [Google Scholar] [CrossRef]
- Das, S.; Brown, S.; Mayes, R.T.; Janke, C.J.; Tsouris, C.; Kuo, L.-J.; Gill, G.; Dai, S. Novel poly(imide dioxime) sorbents: Development and testing for enhanced extraction of uranium from natural seawater. Chem. Eng. J. 2016, 298, 125–135. [Google Scholar] [CrossRef]
- Oyola, Y.; Janke, C.J.; Dai, S. Synthesis, Development, and Testing of High-Surface-Area Polymer-Based Adsorbents for the Selective Recovery of Uranium from Seawater. Ind. Eng. Chem. Res. 2016, 55, 4149–4160. [Google Scholar] [CrossRef]
- Kuo, L.-J.; Janke, C.J.; Wood, J.R.; Strivens, J.E.; Das, S.; Oyola, Y.; Mayes, R.T.; Gill, G.A. Characterization and Testing of Amidoxime-Based Adsorbent Materials to Extract Uranium from Natural Seawater. Ind. Eng. Chem. Res. 2016, 55, 4285–4293. [Google Scholar] [CrossRef]
- Wu, F.; Pu, N.; Ye, G.; Sun, T.; Wang, Z.; Song, Y.; Wang, W.; Huo, X.; Lu, Y.; Chen, J. Performance and Mechanism of Uranium Adsorption from Seawater to Poly(dopamine)-Inspired Sorbents. Environ. Sci. Technol. 2017, 51, 4606–4614. [Google Scholar] [CrossRef]
- Bliznyuk, V.N.; Kołacińska, K.; Pud, A.A.; Ogurtsov, N.A.; Noskov, Y.V.; Powell, B.A.; DeVol, T.A. High effectiveness of pure polydopamine in extraction of uranium and plutonium from groundwater and seawater. RSC Adv. 2019, 9, 30052–30063. [Google Scholar] [CrossRef]
- Shinkai, S.; Kawaguchi, H.; Manabe, O. Selective adsorption of UO22+ to a polymer resin immobilizing calixarene-based uranophiles. J. Polym. Sci. Part C Polym. Lett. 1988, 26, 391–396. [Google Scholar] [CrossRef]
- Metilda, P.; Gladis, J.M.; Venkateswaran, G.; Prasada Rao, T. Investigation of the role of chelating ligand in the synthesis of ion-imprinted polymeric resins on the selective enrichment of uranium(VI). Anal. Chim. Acta 2007, 587, 263–271. [Google Scholar] [CrossRef]
- Mossand, G.; Lelong, E.; Xing, C.; Ndebulia Watchou, F.; Leydier, A.; Arrachart, G.; Pellet-Rostaing, S. Bis-Catecholamide-Based Materials for Uranium Extraction. ChemPlusChem 2023, 88, e202200412. [Google Scholar] [CrossRef]
- Turgis, R.; Leydier, A.; Arrachart, G.; Burdet, F.; Dourdain, S.; Bernier, G.; Miguirditchian, M.; Pellet-Rostaing, S. Uranium Extraction from Phosphoric Acid Using Bifunctional Amido-Phosphonic Acid Ligands. Solvent Extr. Ion Exch. 2014, 32, 478–491. [Google Scholar] [CrossRef]
- He, Y.; Bao, W.; Li, B.; Fu, X.; Na, B.; Yuan, D. Highly Efficient Removal of Uranium from Aqueous Solution by a Novel Robust Phosphonic Acid Functionalized Aromatic-Based Hyper-Crosslinked Porous Polymer. J. Radioanal. Nucl. Chem. 2022, 331, 3745–3756. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, P.; He, T.; Huang, J.; Zhang, S.; Liu, Y.; Wang, Y.; Meng, C.; Yuan, D. Highly Efficient Removal of Uranium from an Aqueous Solution by a Novel Phosphonic Acid-Functionalized Magnetic Microsphere Adsorbent. Int. J. Mol. Sci. 2022, 23, 16227. [Google Scholar] [CrossRef] [PubMed]
- Arrambide, C.; Arrachart, G.; Berthalon, S.; Wehbie, M.; Pellet-Rostaing, S. Extraction and Recovery of Rare Earths by Chelating Phenolic Copolymers Bearing Diglycolamic Acid or Diglycolamide Moieties. React. Funct. Polym. 2019, 142, 147–158. [Google Scholar] [CrossRef]
- Granado, L.; Tavernier, R.; Henry, S.; Auke, R.O.; Foyer, G.; David, G.; Caillol, S. Toward Sustainable Phenolic Thermosets with High Thermal Performances. ACS Sustain. Chem. Eng. 2019, 7, 7209–7217. [Google Scholar]
- Azizian, S. Kinetic models of sorption: A theoretical analysis. J. Colloid Interface Sci. 2004, 276, 47–52. [Google Scholar] [CrossRef]
- Simonin, J.-P. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics. Chem. Eng. J. 2016, 300, 254–263. [Google Scholar] [CrossRef]
- Ho, Y. Citation review of Lagergren kinetic rate equation on adsorption reactions. Scientometrics 2004, 59, 171–177. [Google Scholar]
- Revellame, E.D.; Fortela, D.L.; Sharp, W.; Hernandez, R.; Zappi, M.E. Adsorption kinetic modeling using pseudo-first order and pseudo-second order rate laws: A review. Clean. Eng. Technol. 2020, 1, 100032. [Google Scholar] [CrossRef]
- Cheung, C.W.; Porter, J.F.; McKay, G. Sorption kinetics for the removal of copper and zinc from effluents using bone char. Sep. Purif. Technol. 2000, 19, 55–64. [Google Scholar] [CrossRef]
- Das, A.; Chandrakumar, K.R.S.; Paul, B.; Chopade, S.M.; Majumdar, S.; Singh, A.K.; Kain, V. Enhanced adsorption and separation of zirconium and hafnium under mild conditions by phosphoric acid based ligand functionalized silica gels: Insights from experimental and theoretical investigations. Sep. Purif. Technol. 2020, 239, 116518. [Google Scholar] [CrossRef]
- Trieu, Q.A.; Pellet-Rostaing, S.; Arrachart, G.; Traore, Y.; Kimbel, S.; Daniele, S. Interfacial study of surface-modified ZrO2 nanoparticles with thioctic acid for the selective recovery of palladium and gold from electronic industrial wastewater. Sep. Purif. Technol. 2020, 237, 116353. [Google Scholar] [CrossRef]
- Ho, Y.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Vadivelan, V.; Kumar, K.V. Equilibrium, kinetics, mechanism, and process design for the sorption of methylene blue onto rice husk. J. Colloid Interface Sci. 2005, 286, 90–100. [Google Scholar] [CrossRef]
- Plazinski, W.; Dziuba, J.; Rudzinski, W. Modeling of sorption kinetics: The pseudo-second order equation and the sorbate intraparticle diffusivity. Adsorption 2013, 19, 1055–1064. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Ghosal, P.S.; Gupta, A.K. Determination of thermodynamic parameters from Langmuir isotherm constant-revisited. J. Mol. Liq. 2017, 225, 137–146. [Google Scholar] [CrossRef]
- Limousin, G.; Gaudet, J.-P.; Charlet, L.; Szenknect, S.; Barthès, V.; Krimissa, M. Sorption isotherms: A review on physical bases, modeling and measurement. Appl. Geochem. 2007, 22, 249–275. [Google Scholar] [CrossRef]
- Freundlich, H. Über die Adsorption in Lösungen. Z. Phys. Chem. 1907, 57, 385–470. [Google Scholar] [CrossRef]
- Oye Auke, R.; Arrachart, G.; Tavernier, R.; David, G.; Pellet-Rostaing, S. Terephthalaldehyde–Phenolic Resins as a Solid-Phase Extraction System for the Recovery of Rare-Earth Elements. Polymers 2022, 14, 311. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resin | Chelating Monomer | Chelating Monomer Ratio | Resorcinol Ratio | Resin Form |
|---|---|---|---|---|
| R100 | - | - | 1 | -OH/-ONa |
| TyrAMeP34 | TyrAMeP | 0.34 | 0.66 | -OH/-ONa |
| TyrAMe(Me)P34 | TyrAMe(Me)P | 0.34 | 0.66 | -OH/-ONa |
| TyrAMe(Ph)P34 | TyrAMe(Ph)P | 0.34 | 0.66 | -OH/-ONa |
| TyrAMeP66 | TyrAMeP | 0.66 | 0.34 | -OH |
| TyrAMe(Me)P66 | TyrAMe(Me)P | 0.66 | 0.34 | -OH |
| TyrAMe(Ph)P66 | TyrAMe(Ph)P | 0.66 | 0.34 | -OH |
| Resin | %theoretical | %measured | R |
|---|---|---|---|
| TyrAMeP34 | 29 | H | |
| TyrAMe(Me)P34 | 34 | 34 | Me |
| TyrAMe(Ph)P34 | 37 | Ph | |
| TyrAMeP66 | 41 | H | |
| TyrAMe(Me)P66 | 66 | 49 | Me |
| TyrAMe(Ph)P66 | 53 | Ph |
| EU (%) | EZn (%) | ECu (%) | EV (%) | SFU/Zn | SFU/Cu | SFU/V | |
|---|---|---|---|---|---|---|---|
| TyrAMeP34 | 94 | 17 | 55 | 85 | 76 | 13 | 3 |
| TyrAMe(Me)P34 | 92 | 15 | 53 | 83 | 62 | 10 | 2 |
| TyrAMe(Ph)P34 | 95 | 13 | 62 | 88 | 125 | 11 | 3 |
| TyrAMeP66 | 98 | 18 | 58 | 96 | 295 | 46 | 3 |
| TyrAMe(Me)P66 | 99 | 19 | 62 | 97 | 359 | 51 | 3 |
| TyrAMe(Ph)P66 | 98 | 16 | 21 | 94 | 273 | 32 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lelong, E.; El Khoueiry, C.; Giusti, F.; Arrachart, G.; Pellet-Rostaing, S. Uranium Extraction from Salt Water Using Formo-Phenolic Resin Containing Amido-β-phosphonic Acid Chelating Moiety. Water 2025, 17, 1067. https://doi.org/10.3390/w17071067
Lelong E, El Khoueiry C, Giusti F, Arrachart G, Pellet-Rostaing S. Uranium Extraction from Salt Water Using Formo-Phenolic Resin Containing Amido-β-phosphonic Acid Chelating Moiety. Water. 2025; 17(7):1067. https://doi.org/10.3390/w17071067
Chicago/Turabian StyleLelong, Evan, Claudine El Khoueiry, Fabrice Giusti, Guilhem Arrachart, and Stéphane Pellet-Rostaing. 2025. "Uranium Extraction from Salt Water Using Formo-Phenolic Resin Containing Amido-β-phosphonic Acid Chelating Moiety" Water 17, no. 7: 1067. https://doi.org/10.3390/w17071067
APA StyleLelong, E., El Khoueiry, C., Giusti, F., Arrachart, G., & Pellet-Rostaing, S. (2025). Uranium Extraction from Salt Water Using Formo-Phenolic Resin Containing Amido-β-phosphonic Acid Chelating Moiety. Water, 17(7), 1067. https://doi.org/10.3390/w17071067