
Degradation of Tetracycline Hydrochloride via Activation of Peroxymonosulfate by Magnetic Nickel–Cobalt Ferrite: Role of High-Valent Metal Species as Primary Reactive Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Synthesis of Co0.5Ni0.5Fe2O4 Catalysts
2.3. Characterization of Co0.5Ni0.5Fe2O4 Catalysts
2.4. Degradation Experiments
3. Results and Discussion
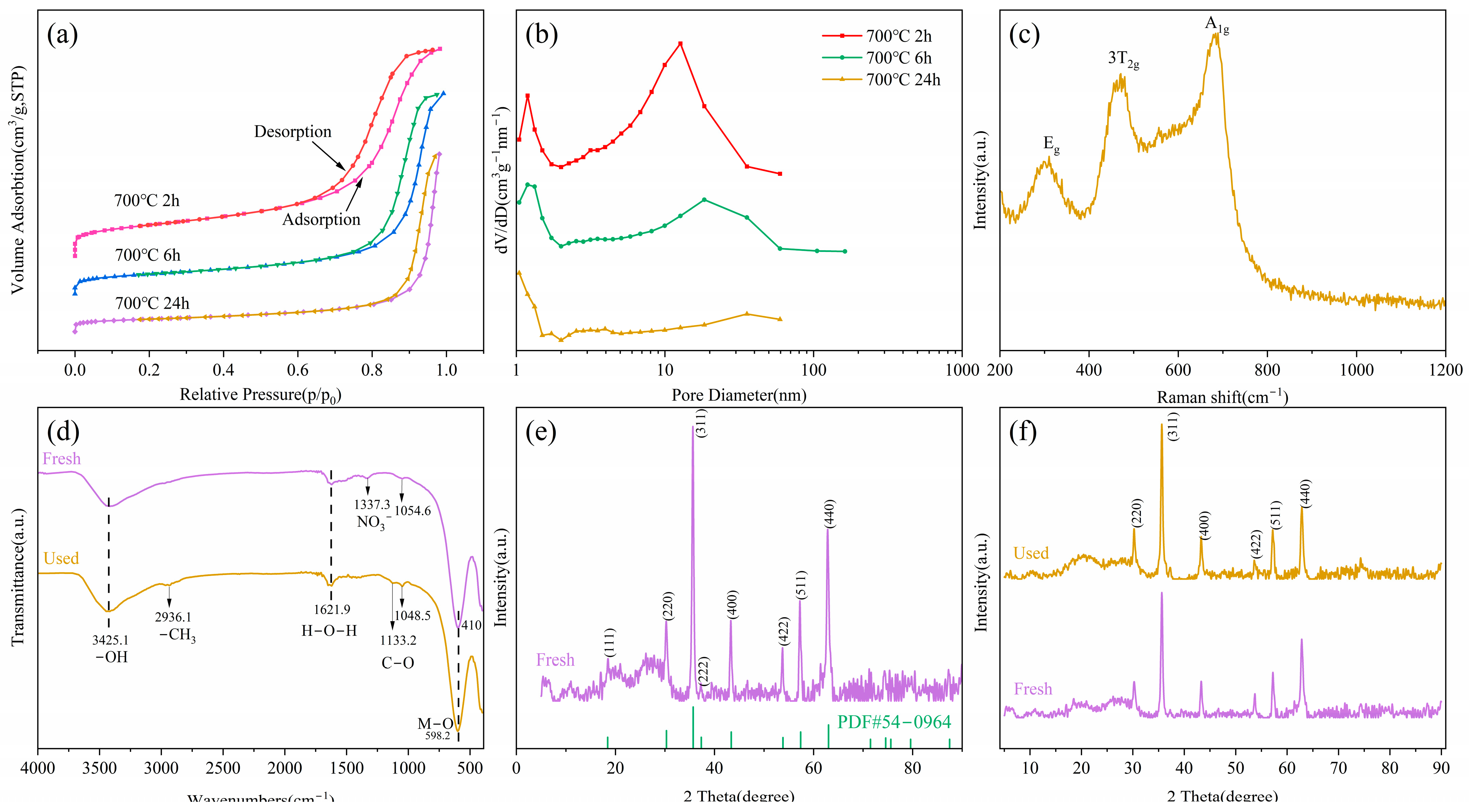
3.1. Characterization of Catalyst
3.2. Catalytic PMS Activation for TCH Degradation
3.3. Identification of Reactive Species
3.3.1. Free Radical Quenching Experiment
3.3.2. EPR Analysis
3.3.3. Evolution Mechanism of
3.4. Reaction Mechanism for PMS Activation by Co0.5Ni0.5Fe2O4 Catalyst
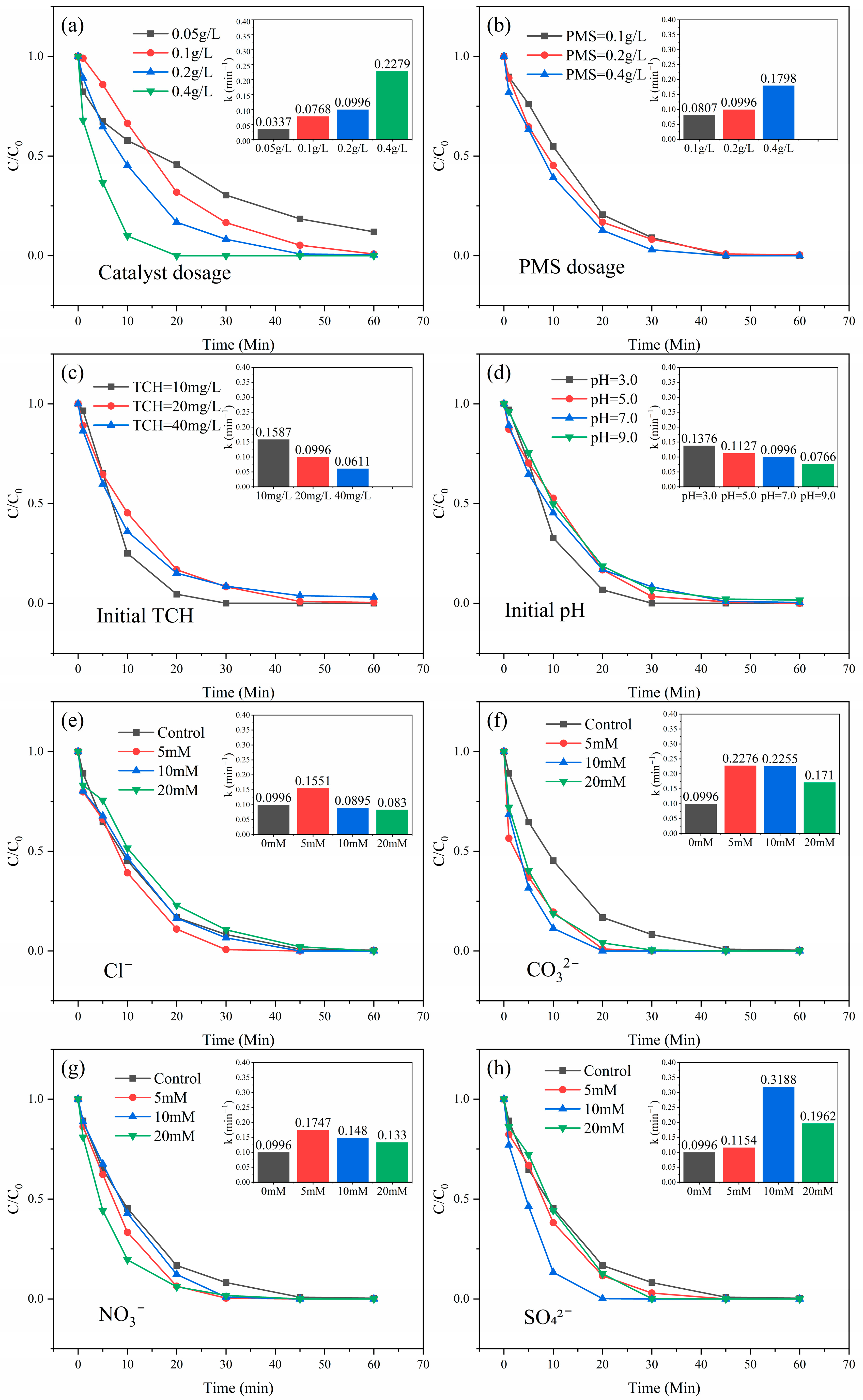
3.5. Influence of Crucial Elements
3.5.1. Influence of Catalyst and PMS Dosage
3.5.2. Influence of Initial Concentration of TCH
3.5.3. Influence of Initial pH
3.5.4. Influence of Co-Existing Ions
3.6. Catalyst Performance Comparison
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Muteeb, G.; Rehman, M.T.; Shahwan, M.; Aatif, M. Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, S.; Liu, Y.; Wang, Y.; Gao, F.; Dai, Y. A review on current pollution and removal methods of tetracycline in soil. Sep. Sci. Technol. 2023, 58, 2578–2602. [Google Scholar] [CrossRef]
- Zhai, W.Y.; Li, M.; Zhang, Q. The mechanism of the synergistic effect of persulfate on the photocatalytic degradation of tetracycline hydrochloride by nano ZnO. Chin. Environ. Sci. 2020, 40, 2483–2492. [Google Scholar]
- Song, Q.; Fang, Y.; Liu, Z.; Li, L.; Wang, Y.; Liang, J.; Huang, Y.; Lin, J.; Hu, L.; Zhang, J.; et al. The performance of porous hexagonal BN in high adsorption capacity towards antibiotic pollutants from aqueous solution. Chem. Eng. J. 2017, 325, 71–79. [Google Scholar] [CrossRef]
- Tang, J.; Wang, J. MOF-derived three-dimensional flower-like FeCu@C composite as an efficient Fenton-like catalyst for sulfamethazine degradation. Chem. Eng. J. 2019, 375, 122007. [Google Scholar] [CrossRef]
- Wu, L.; Sun, Z.; Zhen, Y.; Zhu, S.; Yang, C.; Lu, J.; Tian, Y.; Zhong, D.; Ma, J. Oxygen Vacancy-Induced Nonradical Degradation of Organics: Critical Trigger of Oxygen (O2) in the Fe-Co LDH/Peroxymonosulfate System. Environ. Sci. Technol. 2021, 55, 15400–15411. [Google Scholar] [CrossRef]
- Zheng, X.; Niu, X.; Zhang, D.; Lv, M.; Ye, X.; Ma, J.; Lin, Z.; Fu, M. Metal-based catalysts for persulfate and peroxymonosulfate activation in heterogeneous ways: A review. Chem. Eng. J. 2022, 429, 132323. [Google Scholar] [CrossRef]
- Li, S.; Gong, J. Strategies for improving the performance and stability of Ni-based catalysts for reforming reactions. Chem. Soc. Rev. 2014, 43, 7245–7256. [Google Scholar] [CrossRef]
- Yang, B.; Ma, Q.; Hao, J.; Huang, J.; Wang, Q.; Wang, D.; Zhang, J. Periodate-based advanced oxidation processes: A review focusing on the overlooked role of high-valent iron and manganese species. Chemosphere 2023, 337, 139442. [Google Scholar] [CrossRef] [PubMed]
- Oleg, P.; Andreja, B. Aqueous ferryl(IV) ion: Kinetics of oxygen atom transfer to substrates and oxo exchange with solvent water. Inorg. Chem. 2006, 45, 814–820. [Google Scholar]
- Pestovsky, O.; Stoian, S.; Bominaar, E.L.; Shan, X.; Muenck, E.; Que, L., Jr.; Bakac, A. Aqueous FeIV==O: Spectroscopic identification and oxo-group exchange. Angew. Chem. 2005, 44, 6871–6874. [Google Scholar] [CrossRef]
- Bao, Y.; Lian, C.; Huang, K.; Yu, H.; Liu, W.; Zhang, J.; Xing, M. Generating High-valent Iron-oxo≡ Fe(IV)= O Complexes in Neutral Microenvironments through Peroxymonosulfate Activation by Zn− Fe Layered Double Hydroxides. Angew. Chem. 2022, 134, e202209542. [Google Scholar] [CrossRef]
- Dai, H.; Zhou, W.; Wang, W. Co/N co-doped carbonaceous polyhedron as efficient peroxymonosulfate activator for degradation of organic pollutants: Role of cobalt. Chem. Eng. J. 2021, 417, 127921. [Google Scholar] [CrossRef]
- Larson, V.A.; Battistella, B.; Ray, K.; Lehnert, N.; Nam, W. Iron and manganese oxo complexes, oxo wall and beyond. Nat. Rev. Chem. 2020, 4, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, B.; Dikshit, A.K.; Kim, T.Y.; Tripathi, K.M. Research progress and prospects of spinel ferrite nanostructures for the removal of nitroaromatics from wastewater. ACS Appl. Nano Mater. 2022, 5, 16000–16026. [Google Scholar] [CrossRef]
- Donatos, M.; Kleopatra, M.; Ioannis, K. Perovskite and Spinel Catalysts for Sulfate Radical-Based Advanced Oxidation of Organic Pollutants in Water and Wastewater Systems. Catalysts 2020, 10, 1299. [Google Scholar] [CrossRef]
- Lu, M.; Kang, G.; Deng, Y. Construction of mesoporous S-doped Co 3 O 4 with abundant oxygen vacancies as an efficient activator of PMS for organic dye degradation. CrystEngComm 2023, 25, 2767–2777. [Google Scholar] [CrossRef]
- Lingyun, J.; Wenhan, Y.; Tong, W.; Jingquan, W.; Xiuqin, K.; Suyun, L.; Xinyong, L.; Rui, Q.; Hao, Z. Porous boron nitride micro-nanotubes efficiently anchor CoFe2O4 as a magnetic recyclable catalyst for peroxymonosulfate activation and oxytetracycline rapid degradation. Sep. Purif. Technol. 2022, 290, 120925. [Google Scholar]
- Li, X.; Zhao, Z.; Li, H.; Qian, J. Degradation of organic contaminants in the CoFe2O4/peroxymonosulfate process: The overlooked role of Co (II)-PMS complex. Chem. Eng. J. Adv. 2021, 8, 100143. [Google Scholar] [CrossRef]
- Nguyen, L.T.T.; Nguyen, H.T.T.; Nguyen, L.T.H.; Duong, A.T.T.; Nguyen, H.Q.; Ngo, V.T.M.; Vu, N.V.; Nguyen, D.T.C.; Tran, T.V. Efficient and recyclable Nd3+-doped CoFe2O4 for boosted visible light-driven photocatalytic degradation of Rhodamine B dye. RSC Adv. 2023, 13, 10650–10656. [Google Scholar] [CrossRef] [PubMed]
- Chen, B. Study on the Preparation and Microwave Absorbing Properties of Nickel Cobalt Ferrite; Hunan University: Changsha, China, 2015. [Google Scholar]
- Xin, Y.; Yu, K.; Zhang, L.; Yang, Y.; Yuan, H.; Li, H.; Wang, L.; Zeng, J. Copper-based plasmonic catalysis: Recent advances and future perspectives. Adv. Mater. 2021, 33, 2008145. [Google Scholar] [CrossRef] [PubMed]
- Baruah, P.K.; Mukherjee, N.; Bhagat, B.; Mukherjee, K. Wet chemical synthesis of cubic spinel ferrites: A review addressing phase formation behavior and nano structuring. Cryst. Growth Des. 2024, 24, 1504–1528. [Google Scholar] [CrossRef]
- Horikawa, T.; Do, D.D.; Nicholson, D. Capillary condensation of adsorbates in porous materials. Adv. Colloid Interface Sci. 2011, 169, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Marconi, U.M.B.; Tarazona, P. Fluids in narrow pores: Adsorption, capillary condensation, and critical points. J. Chem. Phys. 1986, 84, 2376–2399. [Google Scholar] [CrossRef]
- Camargo, J.; Espinosa, A.P.; Zabotto, F.; Ramajo, L.; Castro, M. Magnetoelectric interactions in bismuth sodium-potassium titanate-nickel cobalt ferrite lead-free composite ceramics. J. Alloys Compd. 2020, 826, 154129. [Google Scholar] [CrossRef]
- Patil, S.; Meti, S.; Kanavi, P.S.; Bhajantri, R.F.; Anandalli, M.; Mondal, R.; Karmakar, S.; Muhiuddin, M.; Rahman, M.R.; Chethan, B.K.; et al. A study on solubility of bismuth cations in nickel cobalt ferrite nanoparticles and their influence on dielectric and magnetic properties. Mater. Sci. Eng. B 2023, 296, 116570. [Google Scholar] [CrossRef]
- Nayak, S.; Nayak, S.S.; Kittur, A.A.; Nayak, S.; Joshi, D.R. Synthesis, fabrication, and performance evaluation of lanthanum doped nickel cobalt ferrite electrode for supercapacitors in energy storage applications. J. Energy Storage 2024, 101, 113918. [Google Scholar] [CrossRef]
- Suryanarayana, B.; Ramanjaneyulu, K.; Vemuri, R.; Murali, N.; Parajuli, D.; Mulushoa, S.Y.; Praveen, C.; Anantha, P.R.; Ramakrishna, Y.; Chandramouli, K. Effect of Sm3+ substitution on dc electrical resistivity and magnetic properties of Ni–Co ferrites. J. Indian Chem. Soc. 2022, 99, 100623. [Google Scholar] [CrossRef]
- Silva, V.D.; Nascimento, E.P.; Grilo, J.P.F.; Simões, T.A.; Menezes, R.R.; Macedo, D.A.; Medeiros, E.S. Effect of two-step calcination on the formation of nickel oxide hollow nanofibers. Open Ceram. 2021, 5, 100087. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Creating and mastering nano-objects to design advanced catalytic materials. Coord. Chem. Rev. 2011, 255, 1480–1498. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, W.; Wang, W.; Cao, Y.; Fan, G.; Huang, Y.; Qi, M. A comprehensive review of the electrochemical advanced oxidation processes: Detection of free radical, Electrode materials and Application. J. Environ. Chem. Eng. 2024, 12, 113778. [Google Scholar] [CrossRef]
- Yang, B.; Liu, H.; Zhang, J. High-valent metals in advanced oxidation processes: A critical review of their identification methods, formation mechanisms, and reactivity performance. Chem. Eng. J. 2023, 460, 141796. [Google Scholar] [CrossRef]
- Yang, Z.; Guan, X.; Xu, J.; Feng, Y.; Mao, Y.; Xu, L.; Chu, H.; Wu, D. Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt (II)-activated peroxymonosulfate process. Environ. Sci. Technol. 2020, 54, 16231–16239. [Google Scholar]
- Zhou, X.; Zhao, Q.; Wang, J.; Chen, Z.; Chen, Z. Nonradical oxidation processes in PMS-based heterogeneous catalytic system: Generation, identification, oxidation characteristics, challenges response and application prospects. Chem. Eng. J. 2021, 410, 128312. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, H.; Sun, J.; Lu, L.; Li, S. Sulfur Vacancies in Pyrite Trigger the Path to Nonradical Singlet Oxygen and Spontaneous Sulfamethoxazole Degradation: Unveiling the Hidden Potential in Sediments. Environ. Sci. Technol. 2024, 58, 6753–6762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, H.; Bu, L.; Zhou, S.; Shi, Z.; Deng, L. Cu0 incorporated cobalt/nitrogen doped carbonaceous frameworks derived from ZIF-67 (Cu@ CoNC) as PMS activator for efficient degradation of naproxen: Direct electron transfer and 1O2 dominated nonradical mechanisms. Chem. Eng. J. 2023, 454, 139989. [Google Scholar] [CrossRef]
- Yang, P.; Li, S.; Liang, X.; An, X.; Liu, D.; Huang, W. Singlet oxygen-dominated activation of peroxymonosulfate by CuO/MXene nanocomposites for efficient decontamination of carbamazepine under high salinity conditions: Performance and singlet oxygen evolution mechanism. Sep. Purif. Technol. 2022, 285, 120288. [Google Scholar] [CrossRef]
- Krishna, D.N.G.; Philip, J. Review on surface-characterization applications of X-ray photoelectron spectroscopy (XPS): Recent developments and challenges. Appl. Surf. Sci. Adv. 2022, 12, 100332. [Google Scholar] [CrossRef]
- You, Y.; Shi, Z.; Li, Y.; Zhao, Z.; He, B.; Cheng, X. Magnetic cobalt ferrite biochar composite as peroxymonosulfate activator for removal of lomefloxacin hydrochloride. Sep. Purif. Technol. 2021, 272, 118889. [Google Scholar] [CrossRef]
- Zuo, J.; Wang, B.; Kang, J.; Yan, P.; Shen, J.; Wang, S.; Fu, D.; Zhu, X.; She, T.; Zhao, S.; et al. Activation of peroxymonosulfate by nanoscaled NiFe2O4 magnetic particles for the degradation of 2, 4-dichlorophenoxyacetic acid in water: Efficiency, mechanism and degradation pathways. Sep. Purif. Technol. 2022, 297, 121459. [Google Scholar] [CrossRef]
- Yu, F.; Xiao, Y.; Tao, L. The activation of PMS for PFOA degradation by adjustable metal stripping Fe/Co/N@ BC catalysts: Degradation performance with single-line oxygen and high-valent metal oxides as the main active substances. Sep. Purif. Technol. 2024, 351, 127589. [Google Scholar] [CrossRef]
- Yang, Z.; Tan, X.; Zhang, C. Sulfidation of Ni-Fe-LDH for enhanced Fenton-like catalysis: Experimental validation and theoretical calculation. Sep. Purif. Technol. 2024, 336, 126320. [Google Scholar] [CrossRef]
- Liu, S.; Yu, X.F.; Peng, Y.; Ding, X.; Cai, H.; Jin, J.; Li, Z.; Tang, H.; Yang, X. Atomically Dispersed Cobalt Anchored on Hollow Tubular Carbon Nitride Mediates Direct Electron Transfer and Oxygen-Related Active Species Path for Activation of Permonosulfate. Inorg. Chem. 2024, 63, 21260–21274. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, S.; Wang, Y.; Lin, Y.; Guan, Q.; Chen, C. Identifying the role of surface hydroxyl on FeOCl in bridging electron transfer toward efficient persulfate activation. Environ. Sci. Technol. 2023, 57, 12922–12930. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, Q.; Zhang, X.; Dong, L.; Yuan, Y.; Zhang, M.; Rao, P.; Gao, N.; Deng, J. CO2(OH)2CO3 and Fe doped Co2(OH)2CO3 prepared as efficient and stable catalysts for activation of PMS: Two systems comparison and key role of high-valent metal-oxo species in mechanism revealing. Sep. Purif. Technol. 2025, 357, 129980. [Google Scholar]
- Liu, Z.; Gao, Z.; Wu, Q. Activation of persulfate by magnetic zirconium-doped manganese ferrite for efficient degradation of tetracycline. Chem. Eng. J. 2021, 423, 130283. [Google Scholar] [CrossRef]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Luo, H.; Zhou, X.; Chen, Q.; Zhou, J. Removal of 2, 4-dichlorophenoxyacetic acid by the boron-nitrogen co-doped carbon nanotubes: Insights into peroxymonosulfate adsorption and activation. Sep. Purif. Technol. 2021, 259, 118196. [Google Scholar] [CrossRef]
- Mártire, D.O.; Caregnato, P.; Furlong, J.; Allegretti, P.; Gonzalez, M.C. Kinetic study of the reactions of oxoiron (IV) with aromatic substrates in aqueous solutions. Int. J. Chem. Kinet. 2002, 34, 488–494. [Google Scholar] [CrossRef]
- Liu, B.; Guo, W.; Jia, W.; Wang, H.; Zheng, S.; Si, Q.; Zhao, Q.; Luo, H.; Jiang, J.; Ren, N. Insights into the oxidation of organic contaminants by Co (II) activated peracetic acid: The overlooked role of high-valent cobalt-oxo species. Water Res. 2021, 201, 117313. [Google Scholar] [CrossRef]
- Lu, H.; Sui, M.; Yuan, B.; Wang, J.; Lv, Y. Efficient degradation of nitrobenzene by Cu-Co-Fe-LDH catalyzed peroxymonosulfate to produce hydroxyl radicals. Chem. Eng. J. 2019, 357, 140–149. [Google Scholar] [CrossRef]
- Zhou, H.; He, Y.L.; Peng, J.; Duan, X.G.; Lu, X.H.; Zhang, H.; Liu, Y.; He, C.S.; Xiong, Z.K.; Ma, T.Y.; et al. High-valent metal-oxo species transformation and regulation by co-existing chloride: Reaction pathways and impacts on the generation of chlorinated by-products. Water Res. 2024, 257, 121715. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhang, Y.; Fan, Z.; Liu, F.; Li, A. Carbonate-enhanced catalytic activity and stability of Co3O4 nanowires for 1O2-driven bisphenol A degradation via peroxymonosulfate activation: Critical roles of electron and proton acceptors. J. Hazard. Mater. 2020, 393, 122395. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Liu, L.; Xu, J.; Jin, T.; Fu, L.; Pan, Y. Effect of anions and cations on tartrazine removal by the zero-valent iron/peroxymonosulfate process: Efficiency and major radicals. Catalysts 2022, 12, 1114. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Ding, M.; Xu, H. Degradation of Tetracycline Hydrochloride via Activation of Peroxymonosulfate by Magnetic Nickel–Cobalt Ferrite: Role of High-Valent Metal Species as Primary Reactive Agents. Water 2025, 17, 616. https://doi.org/10.3390/w17050616
Xie H, Ding M, Xu H. Degradation of Tetracycline Hydrochloride via Activation of Peroxymonosulfate by Magnetic Nickel–Cobalt Ferrite: Role of High-Valent Metal Species as Primary Reactive Agents. Water. 2025; 17(5):616. https://doi.org/10.3390/w17050616
Chicago/Turabian StyleXie, Hangang, Mingmei Ding, and Hang Xu. 2025. "Degradation of Tetracycline Hydrochloride via Activation of Peroxymonosulfate by Magnetic Nickel–Cobalt Ferrite: Role of High-Valent Metal Species as Primary Reactive Agents" Water 17, no. 5: 616. https://doi.org/10.3390/w17050616
APA StyleXie, H., Ding, M., & Xu, H. (2025). Degradation of Tetracycline Hydrochloride via Activation of Peroxymonosulfate by Magnetic Nickel–Cobalt Ferrite: Role of High-Valent Metal Species as Primary Reactive Agents. Water, 17(5), 616. https://doi.org/10.3390/w17050616