
Metagenomic Characterization of Microbial Pollutants and Antibiotic- and Metal-Resistance Genes in Sediments from the Canals of Venice


Abstract
:1. Introduction
2. Materials and Methods
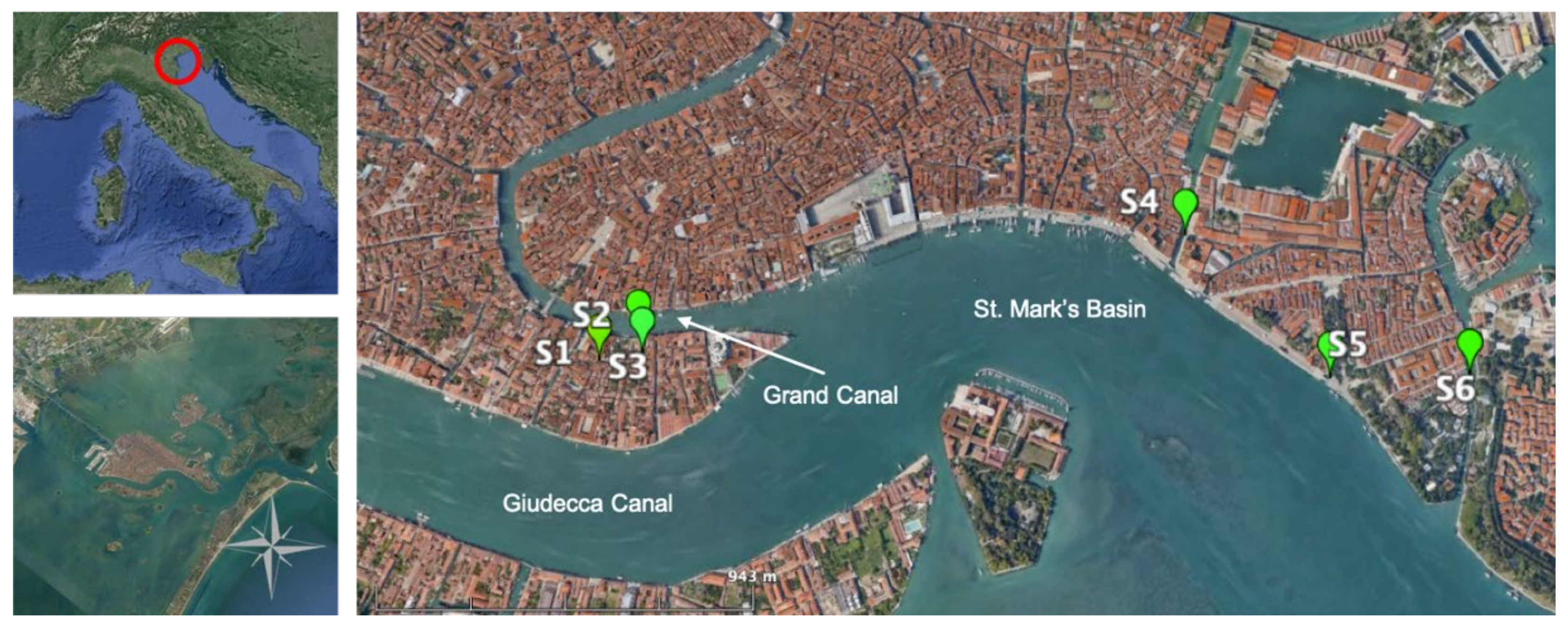
2.1. Study Site
2.2. Sampling Activities
2.3. DNA Extraction and Shotgun Metagenomic Sequencing
2.4. Data Analysis
3. Results and Discussion
3.1. Prokaryotic Diversity and Community Composition
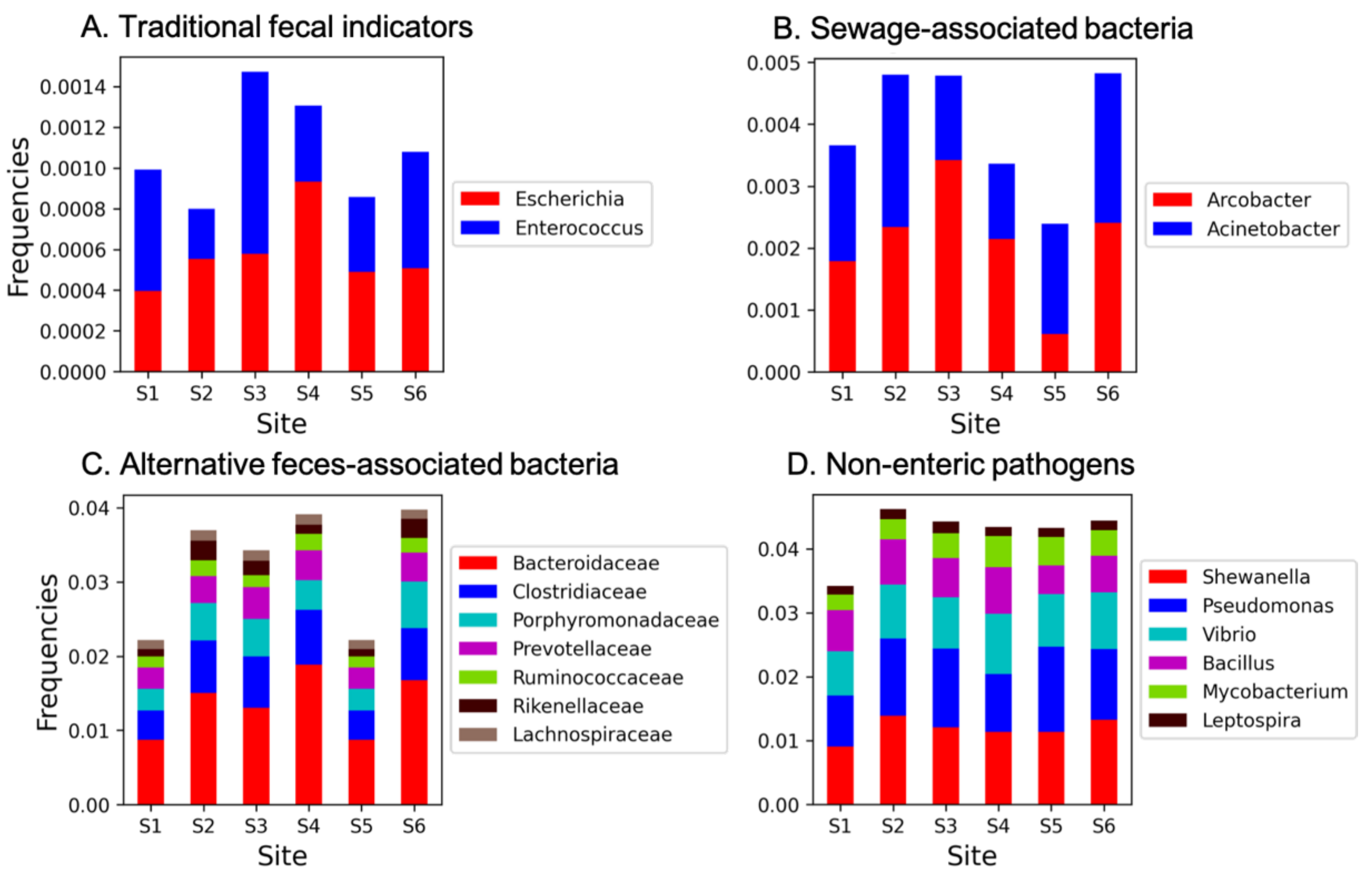
3.2. Microbial Pollutants
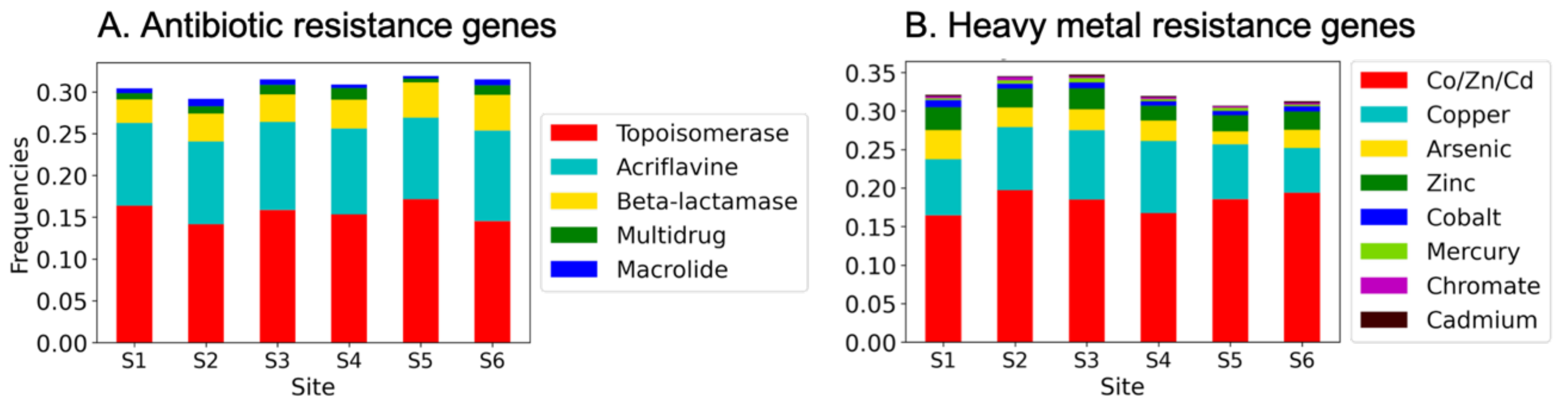
3.3. Antibiotic- and Heavy Metal-Resistance Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fries, J.S.; Characklis, G.W.; Noble, R.T. Attachment of fecal indicator bacteria to particles in the Neuse River Estuary, NC. J. Environ. Eng. 2006, 132, 1338–1345. [Google Scholar] [CrossRef]
- Vörösmarty, C.J.; McIntyre, P.B.; Gessner, M.O.; Dudgeon, D.; Prusevich, A.; Green, P.; Glidden, S.; Bunn, S.E.; Sullivan, C.A.; Liermann, C.R.; et al. Global threats to human water security and river biodiversity. Nature 2010, 67, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.E.; Elliott, K.H. Tracking marine pollution. Science 2013, 340, 556–558. [Google Scholar] [CrossRef]
- Bernhard, A.E.; Goyard, T.; Simonich, M.T.; Field, K.G. Application of a rapid method for identifying fecal pollution sources in a multi-use estuary. Water Res. 2003, 37, 909–913. [Google Scholar] [CrossRef]
- Lipp, E.K.; Kurz, R.; Vincent, R.; Rodriguez-Palacios, C.; Farrah, S.R.; Rose, J.B. The effects of seasonal variability and weather on microbial fecal pollution and enteric pathogens in a subtropical estuary. Estuaries 2001, 24, 266–276. [Google Scholar] [CrossRef]
- Mallin, M.A.; Williams, K.E.; Esham, E.C.; Lowe, R.P. Effect of human development on bacteriological water quality in coastal watersheds. Ecol. Appl. 2000, 10, 1047–1056. [Google Scholar] [CrossRef]
- Milledge, D.G.; Gurjar, S.K.; Bunce, J.T.; Tare, V.; Sinha, R.; Carbonneau, P.E. Population density controls on microbial pollution across the Ganga catchment. Water Res. 2018, 128, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.R.; Gast, R.J.; Fujioka, R.S.; Solo-Gabriele, H.M.; Meschke, J.S.; Amaral-Zettler, L.A.; Del Castillo, E.; Polz, M.F.; Collier, T.K.; Strom, M.S.; et al. The coastal environment and human health: Microbial indicators, pathogens, sentinels and reservoirs. Environ. Health 2008, 7, S3. [Google Scholar] [CrossRef] [Green Version]
- Luna, G.M.; Quero, G.M.; Perini, L. Next generation sequencing reveals distinct fecal pollution signatures in aquatic sediments across gradients of anthropogenic influence. Adv. Oceanogr. Limnol. 2016, 7, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Byappanahalli, M.N.; Nevers, M.B.; Korajkic, A.; Staley, Z.R.; Harwood, V.J. Enterococci in the environment. Microbiol. Mol. Biol. Rev. 2012, 76, 685–706. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Goh, S.G.; Vergara, G.G.R.V.; Fang, H.M.; Rezaeinejad, S.; Chang, S.Y.; Bayen, S.; Lee, W.A.; Sobsey, M.D.; Rose, J.B.; et al. Alternative fecal indicators and their empirical relationships with enteric viruses, Salmonella enterica, and Pseudomonas aeruginosa in surface waters of a tropical urban catchment. Appl. Environ. Microbiol. 2015, 81, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Luna, G.M.; Manini, E.; Turk, V.; Tinta, T.; D’Errico, G.; Baldrighi, E.; Baljak, V.; Buda, D.; Cabrini, M.; Campanelli, A.; et al. Status of faecal pollution in ports: A basin-wide investigation in the Adriatic Sea. Mar. Pollut. Bull. 2019, 147, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Perini, L.; Quero, G.M.; Serrano García, E.; Luna, G.M. Distribution of Escherichia coli in a coastal lagoon (Venice, Italy): Temporal patterns, genetic diversity and the role of tidal forcing. Water Res. 2015, 87, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Quero, G.M.; Fasolato, L.; Vignaroli, C.; Luna, G.M. Understanding the association of Escherichia coli with diverse macroalgae in the lagoon of Venice. Sci. Rep. 2015, 5, 10969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, K.G.; Samadpour, M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Res. 2007, 41, 3517–3538. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Solo-Gabriele, H.M.; Fleming, L.E.; Elmir, S. Monitoring marine recreational water quality using multiple microbial indicators in an urban tropical environment. Water Res. 2004, 38, 3119–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Walk, S.T.; Gordon, D.M.; Feldgarden, M.; Tiedje, J.M.; Konstantinidis, K.T. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. USA 2011, 108, 7200–7205. [Google Scholar] [CrossRef] [Green Version]
- McLellan, S.L.; Newton, R.J.; Vandewalle, J.L.; Shanks, O.C.; Huse, S.M.; Eren, A.M.; Sogin, M.L. Sewage reflects the distribution of human faecal Lachnospiraceae. Environ. Microbiol. 2013, 15, 2213–2227. [Google Scholar] [CrossRef] [Green Version]
- McLellan, S.L.; Eren, A.M. Discovering new indicators of fecal pollution. Trends Microbiol. 2014, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; VandeWalle, J.L.; Borchardt, M.A.; Gorelick, M.H.; McLellan, S.L. Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an Urban harbor. Appl. Environ. Microbiol. 2011, 77, 6972–6981. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Bootsma, M.J.; Morrison, H.G.; Sogin, M.L.; McLellan, S.L. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of lake Michigan. Microb. Ecol. 2013, 65, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Buccheri, M.A.; Salvo, E.; Coci, M.; Quero, G.M.; Zoccarato, L.; Privitera, V.; Rappazzo, G. Investigating microbial indicators of anthropogenic marine pollution by 16S and 18S High-Throughput Sequencing (HTS) library analysis. FEMS Microbiol. Lett. 2019, 366, fnz179. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Campanelli, A.; Frapiccini, E.; Luna, G.M.; Quero, G.M. Occurrence and distribution of microbial pollutants in coastal areas of the Adriatic Sea influenced by river discharge. Environ. Pollut. 2021, 285, 117672. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Techtmann, S.M.; Zaggia, L.; Luna, G.M.; Quero, G.M. Partitioning and sources of microbial pollution in the Venice Lagoon. Sci. Total Environ. 2021, 818, 151755. [Google Scholar] [CrossRef]
- Ming, H.; Ma, Y.; Gu, Y.; Su, J.; Guo, J.; Li, J.; Li, X.; Jin, Y.; Fan, J. Enterococci may not present the pollution of most enteric pathogenic bacteria in recreational seawaters of Xinghai bathing Beach, China. Ecol. Indicat. 2020, 110, 105938. [Google Scholar] [CrossRef]
- Li, W.; Su, H.; Cao, Y.; Wang, L.; Hu, X.; Xu, W.; Xu, Y.; Li, Z.; Wen, G. Antibiotic resistance genes and bacterial community dynamics in the seawater environment of Dapeng Cove, South China. Sci. Total Environ. 2020, 723, 138027. [Google Scholar] [CrossRef]
- Shao, S.; Hu, Y.; Cheng, J.; Chen, Y. Research progress on distribution, migration, transformation of antibiotics and antibiotic resistance genes (ARGs) in aquatic environment. Crit. Rev. Biotechnol. 2018, 38, 1195–1208. [Google Scholar] [CrossRef]
- Calero-Cáceres, W.; Balcázar, J.L. Antibiotic resistance genes in bacteriophages from diverse marine habitats. Sci. Total Environ. 2019, 654, 452–455. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D.J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Di Cesare, A.; Eckert, E.M.; D’Urso, S.; Bertoni, R.; Gillan, D.C.; Wattiez, R.; Corno, G. Co-occurrence of integrase 1, antibiotic and heavy metal resistance genes in municipal wastewater treatment plants. Water Res. 2016, 94, 208–214. [Google Scholar] [CrossRef]
- Vierheilig, J.; Savio, D.; Farnleitner, A.H.; Reischer, G.H.; Ley, R.E.; Mach, R.L.; Farnleitner, A.H.; Reischer, G.H. Potential applications of next generation DNA sequencing of 16S rRNA gene amplicons in microbial water quality monitoring. Water Sci. Technol. 2015, 72, 1962–1972. [Google Scholar] [CrossRef] [Green Version]
- Rotter, A.; Barbier, M.; Bertoni, F.; Bones, A.M.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Cegłowska, M.; Chirivella-Martorell, J.; Dalay, M.C.; et al. The essentials of marine biotechnology. Front. Mar. Sci. 2021, 8, 158. [Google Scholar] [CrossRef]
- Umgiesser, G.; Canu, D.M.; Cucco, A.; Solidoro, C. A finite element model for the Venice Lagoon. Development, set up, calibration and validation. J. Mar. Syst. 2004, 51, 123–145. [Google Scholar] [CrossRef]
- Cucco, A.; Umgiesser, G. Modelling the Venice lagoon residence time. Ecol. Model. 2006, 193, 34–51. [Google Scholar] [CrossRef]
- Lagi, F.; Corti, G.; Meli, M.; Pinto, A.; Bartoloni, A. Leptospirosis acquired by tourists in Venice, Italy. J. Travel Med. 2013, 20, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Bonello, V.; Faraone, C.; Gambarotto, F.; Nicoletto, L.; Pedrini, G. Clusters in formation in a deindustrialized area: Urban regeneration and structural change in Porto Marghera (Venice). Compet. Rev. 2020, 30, 417–436. [Google Scholar] [CrossRef]
- Scarpa, G.M.; Zaggia, L.; Manfè, G.; Lorenzetti, G.; Parnell, K.; Soomere, T.; Rapaglia, J.; Molinaroli, E. The effects of ship wakes in the Venice Lagoon and implications for the sustainability of shipping in coastal waters. Sci. Rep. 2019, 9, 19014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaggia, L.; Rosso, J.; Zonta, R. Sulphate reduction in the sediment of the Venice canals (Italy). Mar. Pollut. Bull. 2007, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, A.; Facca, C. Annual growth and environmental relationships of the invasive species Sargassum muticum and Undaria pinnatifida in the lagoon of Venice. Estuar. Coast. Shelf Sci. 2013, 129, 162–172. [Google Scholar] [CrossRef]
- Coraci, E.; Umgiesser, G.; Zonta, R. Hydrodynamic and sediment transport 752 modelling in the canals of Venice (Italy). Estuar. Coast. Shelf Sci. 2007, 75, 250–260. [Google Scholar] [CrossRef]
- Madricardo, F.; Foglini, F.; Campiani, E.; Grande, V.; Catenacci, E.; Petrizzo, A.; Kruss, A.; Toso, C.; Trincardi, F. Assessing the human footprint on the sea-floor of coastal systems: The case of the Venice Lagoon, Italy. Sci. Rep. 2019, 9, 6615. [Google Scholar] [CrossRef] [PubMed]
- Ostoich, M.; Ghezzo, M.; Umgiesser, G.; Zambon, M.; Tomiato, L.; Ingegneri, F.; Mezzadri, G. Modelling as decision support for the localisation of submarine urban wastewater outfall: Venice lagoon (Italy) as a case study. Environ. Sci. Pollut. Res. 2018, 25, 34306–34318. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Quero, G.M.; Giovannelli, D.; Manini, E.; Vignaroli, C.; Avio, C.G.; De Marco, R.; Luna, G.M. Major role of surrounding environment in shaping biofilm community composition on marine plastic debris. Front. Mar. Sci. 2020, 7, 262. [Google Scholar] [CrossRef]
- Quero, G.M.; Perini, L.; Pesole, G.; Manzari, C.; Lionetti, C.; Bastianini, M.; Marini, M.; Luna, G.M. Seasonal rather than spatial variability drives planktonic and benthic bacterial diversity in a microtidal lagoon and the adjacent open sea. Mol. Ecol. 2017, 26, 5961–5973. [Google Scholar] [CrossRef]
- Meyer, F.; Bagchi, S.; Chaterji, S.; Gerlach, W.; Grama, A.; Harrison, T.; Paczian, T.; Trimble, W.L.; Wilke, A. MG-RAST version 4—lessons learned from a decade of low-budget ultra-high-throughput metagenome analysis. Brief. Bioinform. 2019, 20, 1151–1159. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Rosso, G.E.; Muday, J.A.; Curran, J.F. Tools for metagenomic analysis at wastewater treatment plants: Application to a foaming episode. Water Environ. Res. 2018, 90, 258–268. [Google Scholar] [CrossRef]
- Magurran, A.E.; McGill, B.J. Biological Diversity: Frontiers in Measurement and Assessment; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Jean, M.R.; Gonzalez-Rizzo, S.; Gauffre-Autelin, P.; Lengger, S.K.; Schouten, S.; Gros, O. Two new Beggiatoa species inhabiting marine mangrove sediments in the Caribbean. PLoS ONE 2015, 10, e0117832. [Google Scholar] [CrossRef] [Green Version]
- Teske, A.; Nelson, D.C. The genera Beggiatoa and Thioploca. In The Prokaryotes, 4th ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 6, pp. 784–810. [Google Scholar]
- Jørgensen, B.B. Distribution of colorless sulfur bacteria (Beggiatoa spp.) in a coastal marine sediment. Mar. Biol. 1977, 41, 19–28. [Google Scholar] [CrossRef]
- Mussmann, M.; Schulz, H.N.; Strotmann, B.; Kjaer, T.; Nielsen, L.P.; Rossello-Mora, R.A.; Aman, R.I.; Jørgensen, B.B. Phylogeny and distribution of nitrate-storing Beggiatoa spp. in coastal marine sediments. Environ. Microbiol. 2003, 5, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Brysch, K.; Schneider, C.; Fuchs, G.; Widdel, F. Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch. Microbiol. 1987, 148, 264–274. [Google Scholar] [CrossRef]
- Yu, S.; Yao, P.; Liu, J.; Zhao, B.; Zhang, G.; Zhao, M.; Yu, Z.; Zhang, X.H. Diversity, abundance, and niche differentiation of ammonia-oxidizing prokaryotes in mud deposits of the eastern China marginal seas. Front. Microbiol. 2016, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.J.; Park, S.J.; Yoon, D.N.; Schouten, S.; Sinninghe Damsté, J.S.; Rhee, S.K. Cultivation of autotrophic ammonia-oxidizing archaea from marine sediments in coculture with sulfur-oxidizing bacteria. Appl. Environ. Microbiol. 2010, 76, 7575–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Huang, X.; Zheng, T.L. Responses of bacterial and archaeal communities to nitrate stimulation after oil pollution in mangrove sediment revealed by Illumina sequencing. Mar. Pollut. Bull. 2016, 109, 281–289. [Google Scholar] [CrossRef]
- Kuypers, M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Preisler, A.; De Beer, D.; Lichtschlag, A.; Lavik, G.; Boetius, A.; Jørgensen, B.B. Biological and chemical sulfide oxidation in a Beggiatoa inhabited marine sediment. ISME J. 2007, 1, 341–353. [Google Scholar] [CrossRef]
- Fattore, E.; Benfenati, E.; Marelli, R.; Cools, E.; Fanelli, R. Sterols in sediment samples from Venice Lagoon, Italy. Chemosphere 1996, 33, 2383–2393. [Google Scholar] [CrossRef]
- O’Mullan, G.D.; Juhl, A.R.; Reichert, R.; Schneider, E.; Martinez, N. Patterns of sediment-associated fecal indicator bacteria in an urban estuary: Benthic-pelagic coupling and implications for shoreline water quality. Sci. Total Environ. 2019, 656, 1168–1177. [Google Scholar] [CrossRef]
- Luna, G.M.; Vignaroli, C.; Rinaldi, C.; Pusceddu, A.; Nicoletti, L.; Gabellini, M.; Danovaro, R.; Biavasco, F. Extraintestinal Escherichia coli carrying virulence genes in coastal marine sediments. Appl. Environ. Microbiol. 2010, 76, 5659–5668. [Google Scholar] [CrossRef] [Green Version]
- Jurelevicius, D.; Cotta, S.R.; Montezzi, L.F.; Dias, A.C.; Mason, O.U.; Picao, R.C.; Janet, K.J.; Seldin, L. Enrichment of potential pathogens in marine microbiomes with different degrees of anthropogenic activity. Environ. Poll. 2021, 268, 115757. [Google Scholar] [CrossRef] [PubMed]
- Satomi, M. The family Shewanellaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: New York, NY, USA, 2014; pp. 597–625. [Google Scholar]
- Janda, J.M. Shewanella: A marine pathogen as an emerging cause of human disease. Clin. Microbiol. Newsl. 2014, 36, 25–29. [Google Scholar] [CrossRef]
- Poirel, L.; Héritier, C.; Nordmann, P. Chromosome-encoded ambler class D β-lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob. Agents Chemother. 2004, 48, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Potron, A.; Poirel, L.; Nordmann, P. Origin of OXA-181, an emerging carbapenem-hydrolyzing oxacillinase, as a chromosomal gene in Shewanella xiamenensis. Antimicrob. Agents Chemother. 2011, 55, 4405–4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousfi, K.; Touati, A.; Lefebvre, B.; Fournier, É.; Côté, J.C.; Soualhine, H.; Walker, M.; Bougdour, D.; Tremblay, C.; Bekal, S. A novel plasmid, pSx1, harboring a new Tn1696 derivative from extensively drug-resistant Shewanella xiamenensis encoding OXA-416. Microb. Drug Resist. 2016, 23, 429–436. [Google Scholar] [CrossRef]
- Baaziz, H.; Lemaire, O.N.; Jourlin-Castelli, C.; Iobbi-Nivol, C.; Méjean, V.; Alatou, R.; Fons, M. Draft genome sequence of Shewanella algidipiscicola H1, a highly chromate-resistant strain isolated from Mediterranean marine sediments. Microbiol. Resour. Announc. 2018, 7, e00905-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.C.; Hugie, C.N.; Kile, M.L.; Navab-Daneshmand, T. Association between heavy metals and antibiotic-resistant human pathogens in environmental reservoirs: A review. Front. Environ. Sci. Eng. 2019, 13, 46. [Google Scholar] [CrossRef]
- Zheng, D.; Yin, G.; Liu, M.; Chen, C.; Jiang, Y.; Hou, L.; Zheng, Y. A systematic review of antibiotics and antibiotic resistance genes in estuarine and coastal environments. Sci. Total Environ. 2021, 777, 146009. [Google Scholar] [CrossRef]
- Vignaroli, C.; Pasquaroli, S.; Citterio, B.; Di Cesare, A.; Mangiaterra, G.; Fattorini, D.; Biavasco, F. Antibiotic and heavy metal resistance in enterococci from coastal marine sediment. Environ. Pollut. 2018, 237, 406–413. [Google Scholar] [CrossRef]
- Vickers, A.A.; Chopra, I.; O’neill, A.J. Intrinsic novobiocin resistance in Staphylococcus saprophyticus. Antimicrob. Agents Chemother. 2007, 51, 4484–4485. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare, A.; Pasquaroli, S.; Vignaroli, C.; Paroncini, P.; Luna, G.M.; Manso, E.; Biavasco, F. The marine environment as a reservoir of enterococci carrying resistance and virulence genes strongly associated with clinical strains. Environ. Microbiol. Rep. 2014, 6, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Eckert, E.M.; Quero, G.M.; Di Cesare, A.; Manfredini, G.; Mapelli, F.; Borin, S.; Fontaneto, D.; Luna, G.M.; Corno, G. Antibiotic disturbance affects aquatic microbial community composition and food web interactions but not community resilience. Mol. Ecol. 2019, 28, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Petrin, S.; Fontaneto, D.; Losasso, C.; Eckert, E.M.; Tassistro, G.; Borello, A.; Ricci, A.; Wilson, W.H.; Pruzzo, C.; et al. ddPCR applied on archived Continuous Plankton Recorder samples reveals long-term occurrence of class 1 integrons and a sulphonamide resistance gene in marine plankton communities. Environ. Microbiol. Rep. 2018, 10, 458–464. [Google Scholar] [CrossRef]
- Su, S.; Li, C.; Yang, J.; Xu, Q.; Qiu, Z.; Xue, B.; Wang, S.; Zhao, C.; Xiao, Z.; Wang, J.; et al. Distribution of antibiotic resistance genes in three different natural water bodies-a lake, river and sea. Int. J. Environ. Res. Public Health 2020, 17, 552. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Wang, H.; Chen, Q.; Su, J.Q.; Gad, M.; Li, J.; Mulla, S.I.; Yu, C.P.; Hu, A. Fecal pollution mediates the dominance of stochastic assembly of antibiotic resistome in an urban lagoon (Yundang lagoon), China. J. Hazard. Mater. 2021, 417, 126083. [Google Scholar] [CrossRef]
- Wang, H.; Hou, L.; Liu, Y.; Liu, K.; Zhang, L.; Huang, F.; Rashid, A.; Hu, A.; Yu, C. Horizontal and vertical gene transfer drive sediment antibiotic resistome in an urban lagoon system. J. Environ. Sci. 2021, 102, 11–23. [Google Scholar] [CrossRef]
- Sabatino, R.; Di Cesare, A.; Dzhembekova, N.; Fontaneto, D.; Eckert, E.M.; Corno, G.; Moncheva, S.; Bertoni, R.; Callieri, C. Spatial distribution of antibiotic and heavy metal resistance genes in the Black Sea. Mar. Pollut. Bull. 2020, 160, 111635. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [Green Version]
- Manna, M.S.; Tamer, Y.T.; Gaszek, I.; Poulides, N.; Ahmed, A.; Wang, X.; Toprak, F.C.R.; Woodard, D.R.; Koh, A.Y.; Williams, N.S.; et al. A trimethoprim derivative impedes antibiotic resistance evolution. Nat. Commun. 2021, 12, 2949. [Google Scholar] [CrossRef]
- Müller, A.; Österlund, H.; Marsalek, J.; Viklander, M. The pollution conveyed by urban runoff: A review of sources. Sci. Total Environ. 2020, 709, 136125. [Google Scholar] [CrossRef] [PubMed]
- Zonta, R.; Zaggia, L.; Collavini, F.; Costa, F.; Scattolin, M. Sediment contamination assessment of the Venice canal network (Italy). In Flooding and Environmental Challenges for Venice and Its Lagoon: State of Knowledge; Fletcher, C., Spencer, T., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 603–615. [Google Scholar]
- Zonta, R.; Cassin, D.; Pini, R.; Dominik, J. Substantial decrease in contaminant concentrations in the sediments of the Venice (Italy) canal network in the last two decades—implications for sediment management. Water 2020, 12, 1965. [Google Scholar] [CrossRef]
- Sfriso, A.; Buosi, A.; Tomio, Y.; Juhmani, A.-S.; Mistri, M.; Munari, C.; Sfriso, A.A. Trends of nitrogen and phosphorus in surface sediments of the lagoons of the Northern Adriatic Sea as a study case. Water 2021, 13, 2914. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Site | Richness | Inverse Simpson | G50 | G95 |
|---|---|---|---|---|
| S1 | 850 | 56.2 | 42 | 421 |
| S2 | 770 | 158 | 57 | 423 |
| S3 | 797 | 148 | 57 | 427 |
| S4 | 707 | 143 | 53 | 398 |
| S5 | 798 | 111 | 56 | 415 |
| S6 | 759 | 156 | 56 | 404 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curran, J.F.; Zaggia, L.; Quero, G.M. Metagenomic Characterization of Microbial Pollutants and Antibiotic- and Metal-Resistance Genes in Sediments from the Canals of Venice. Water 2022, 14, 1161. https://doi.org/10.3390/w14071161
Curran JF, Zaggia L, Quero GM. Metagenomic Characterization of Microbial Pollutants and Antibiotic- and Metal-Resistance Genes in Sediments from the Canals of Venice. Water. 2022; 14(7):1161. https://doi.org/10.3390/w14071161
Chicago/Turabian StyleCurran, James F., Luca Zaggia, and Grazia Marina Quero. 2022. "Metagenomic Characterization of Microbial Pollutants and Antibiotic- and Metal-Resistance Genes in Sediments from the Canals of Venice" Water 14, no. 7: 1161. https://doi.org/10.3390/w14071161
APA StyleCurran, J. F., Zaggia, L., & Quero, G. M. (2022). Metagenomic Characterization of Microbial Pollutants and Antibiotic- and Metal-Resistance Genes in Sediments from the Canals of Venice. Water, 14(7), 1161. https://doi.org/10.3390/w14071161