
An Autonomous Device for Solar Hydrogen Production from Sea Water

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of CoHFe Nanoparticles
2.2. Electrode Preparation
2.3. Electrochemical Characterization
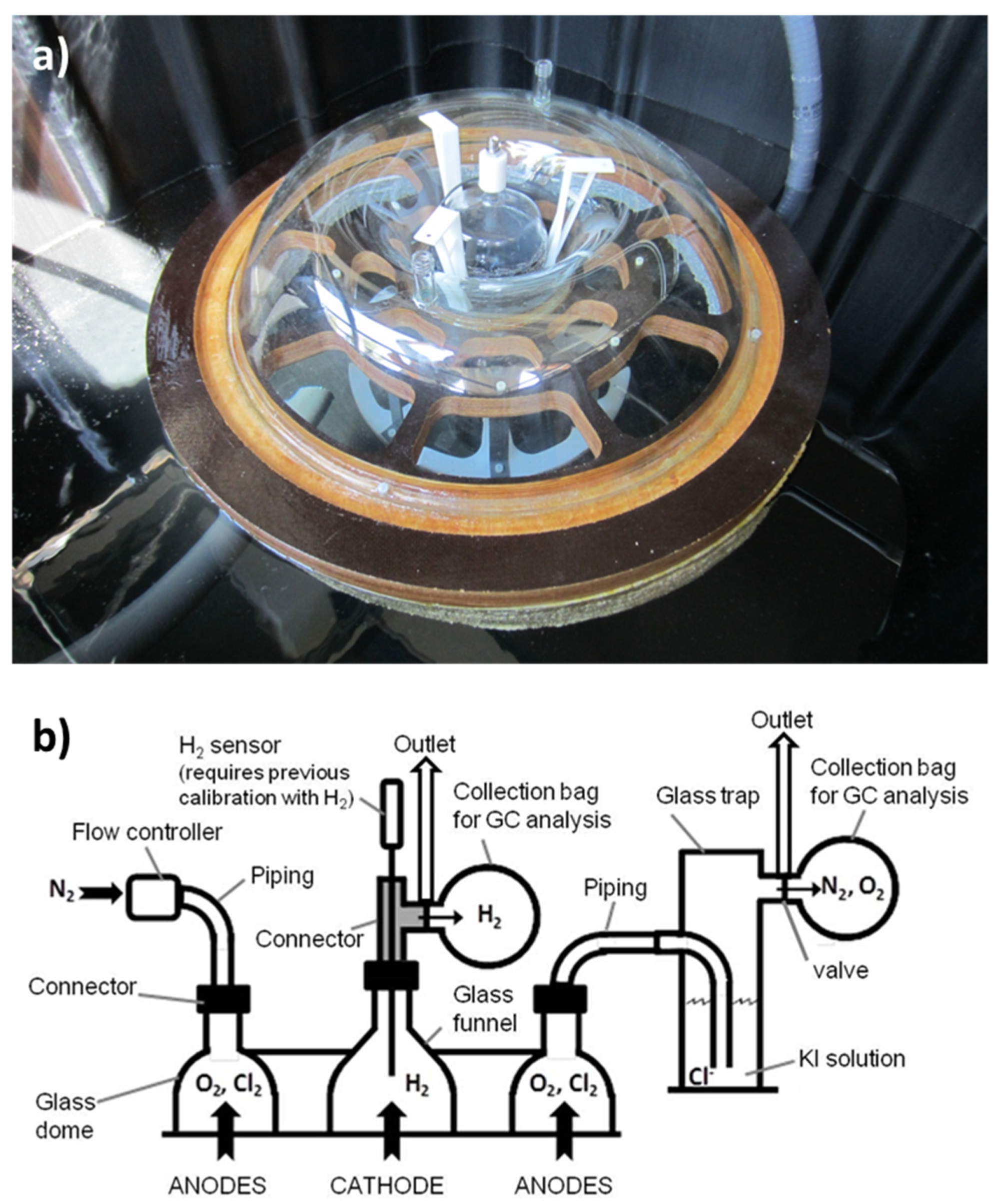
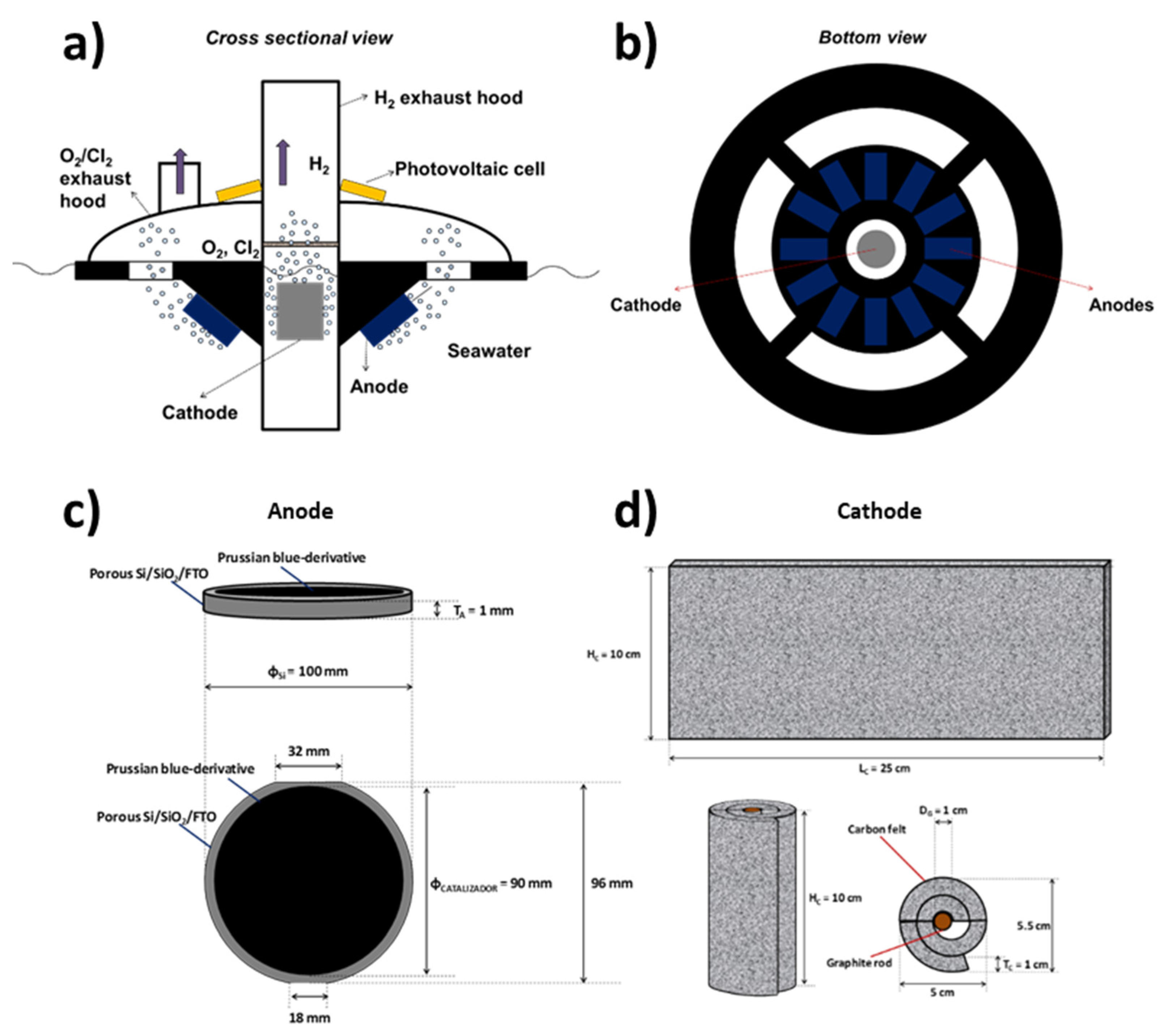
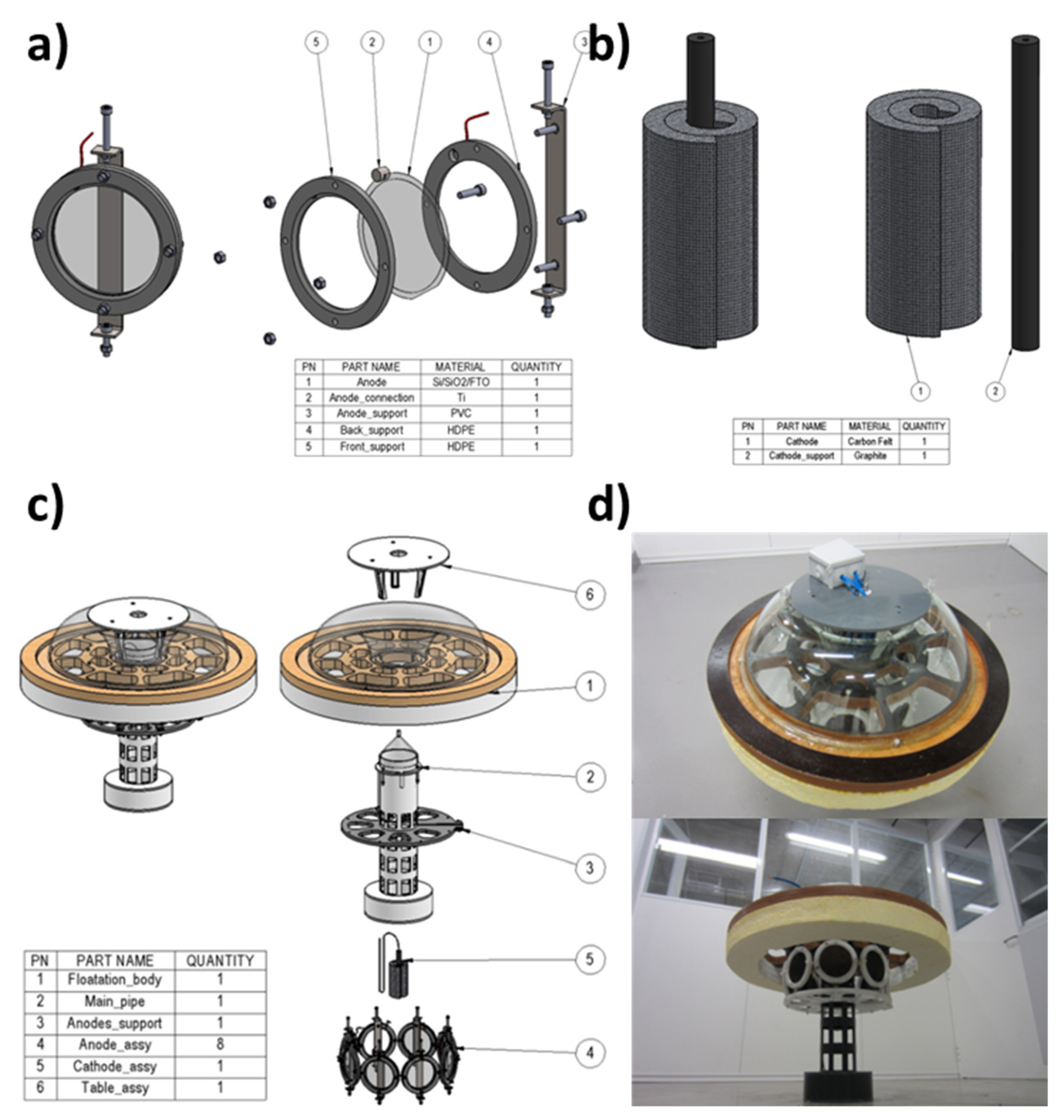
2.4. Seawater Electrolyzer Prototype
3. Results
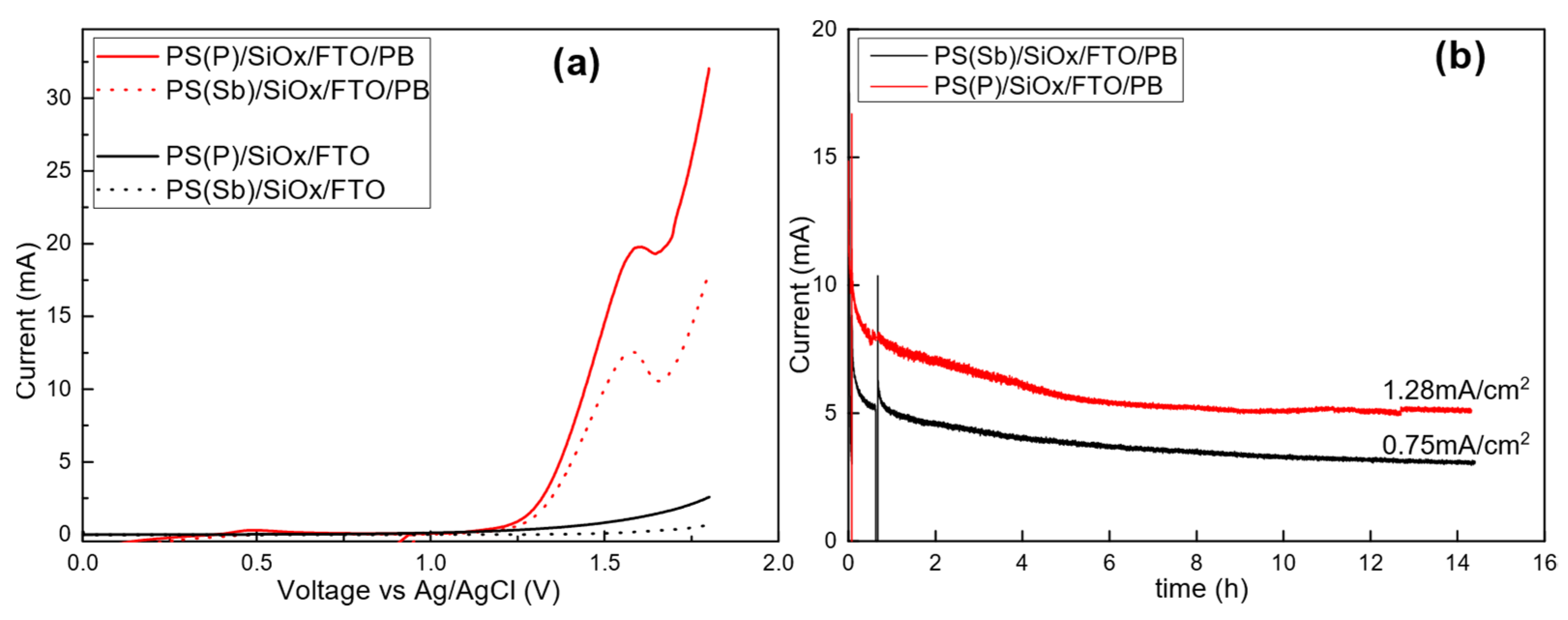
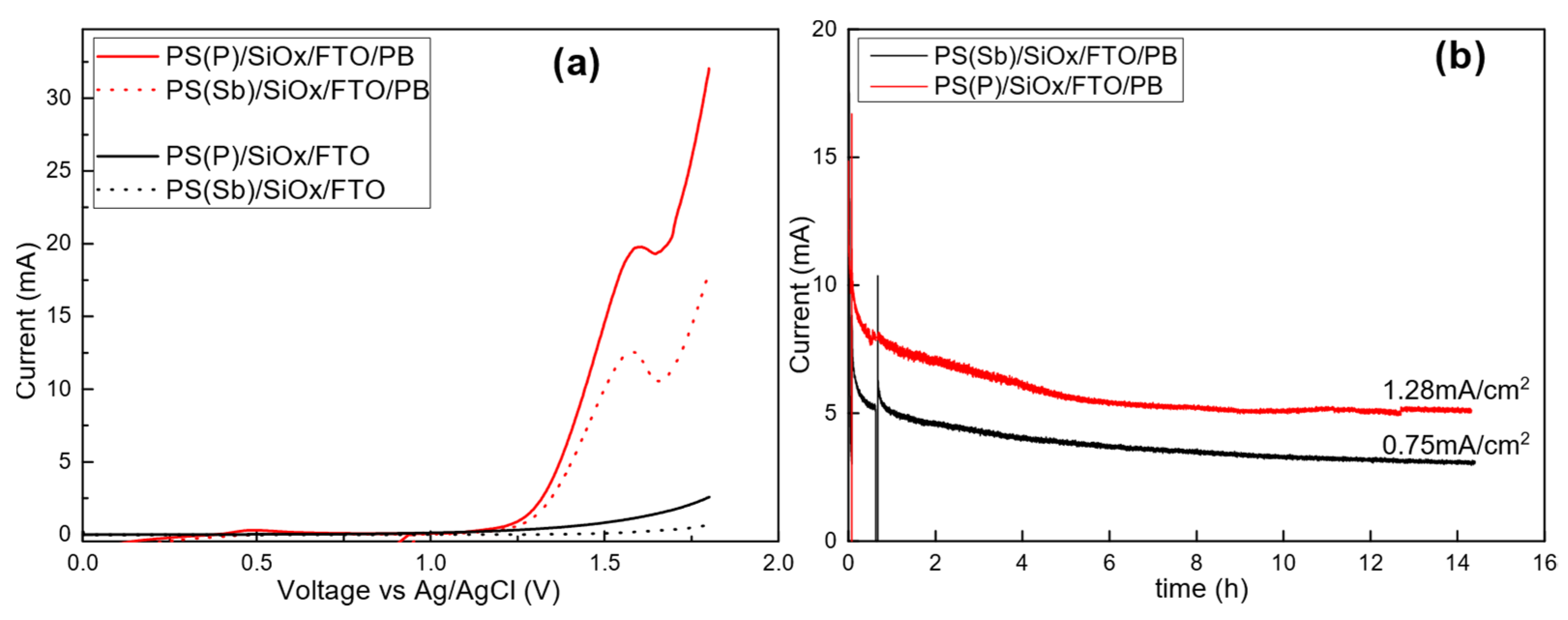
3.1. Anode and Cathode
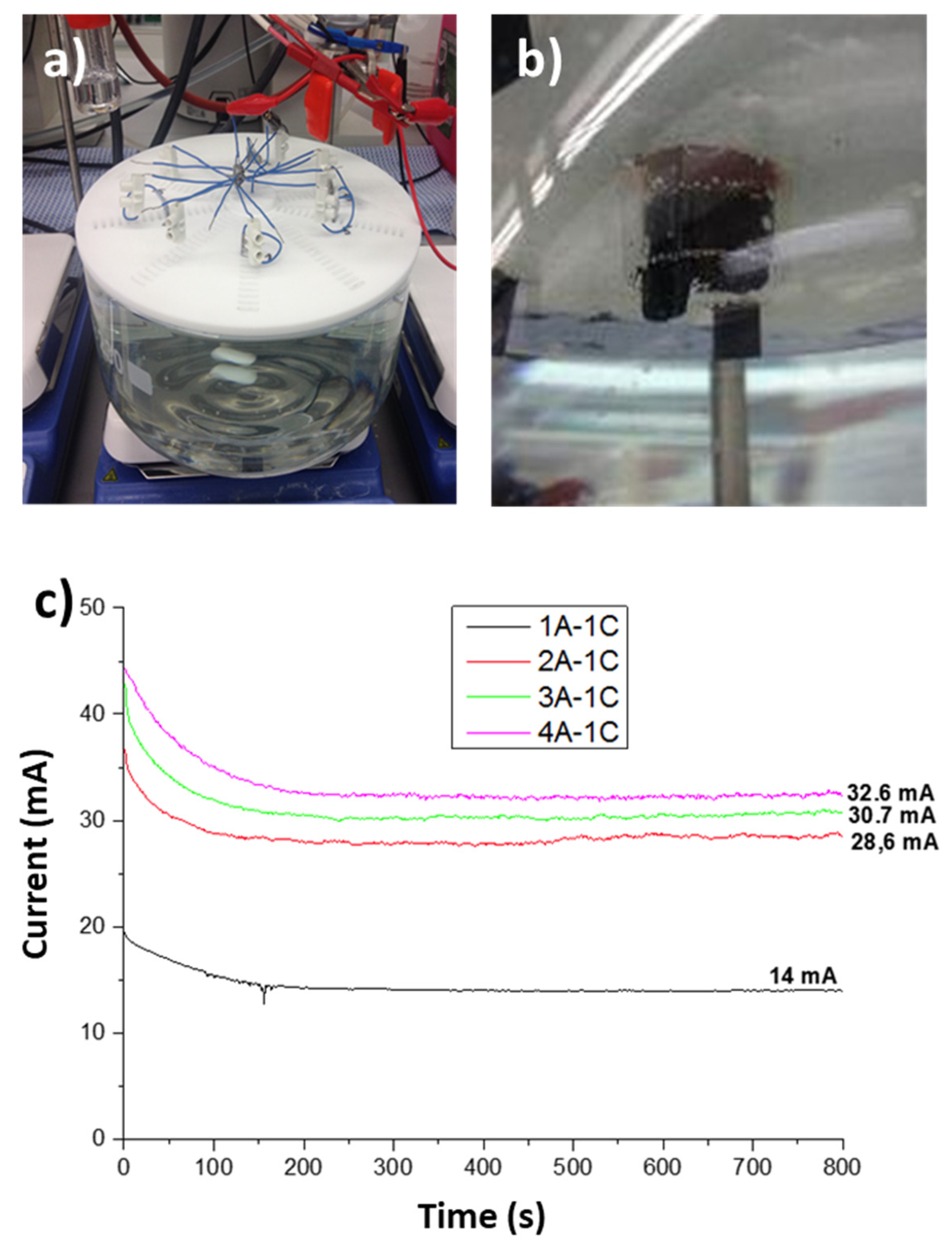
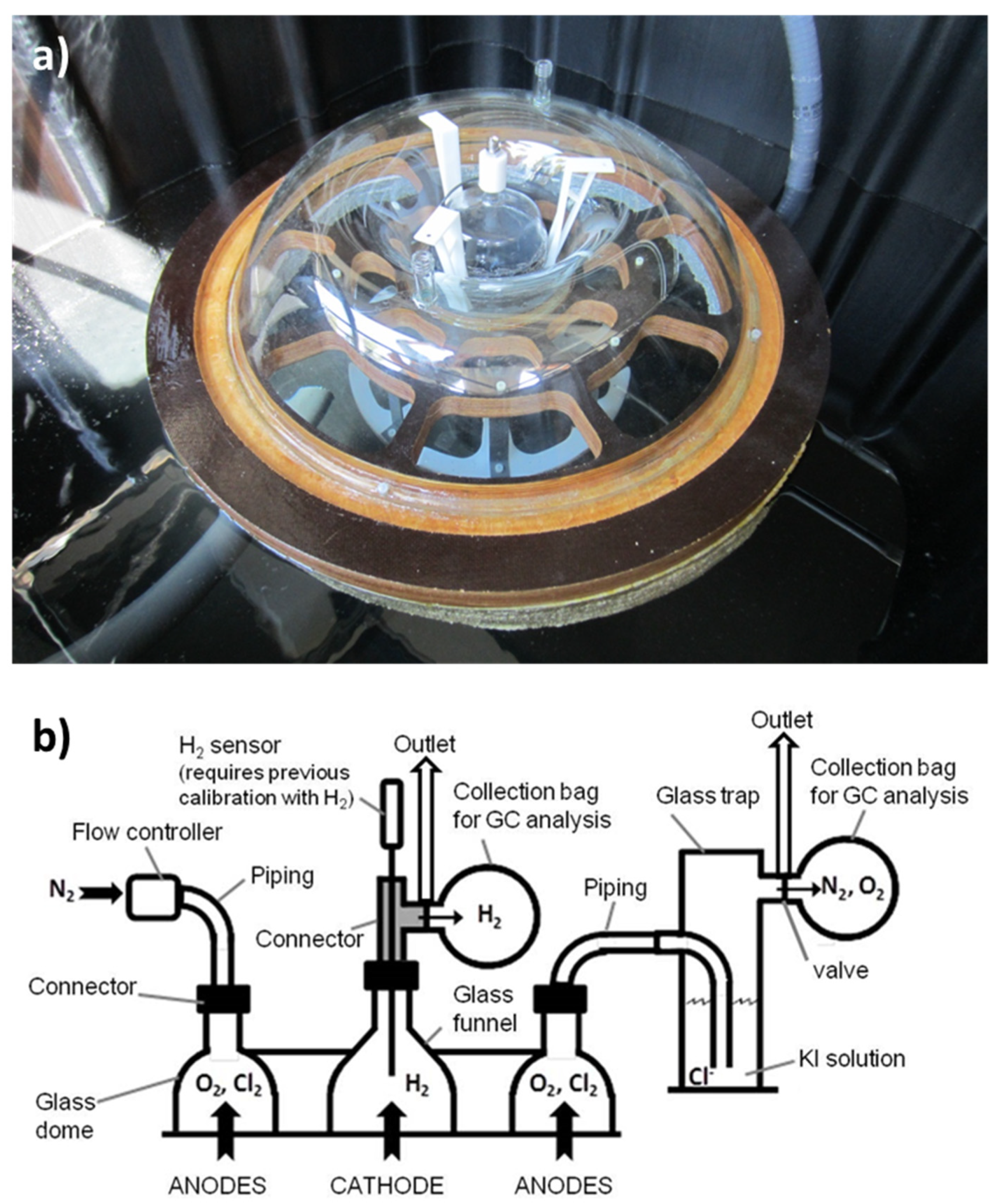
3.2. Prototype Design and Construction
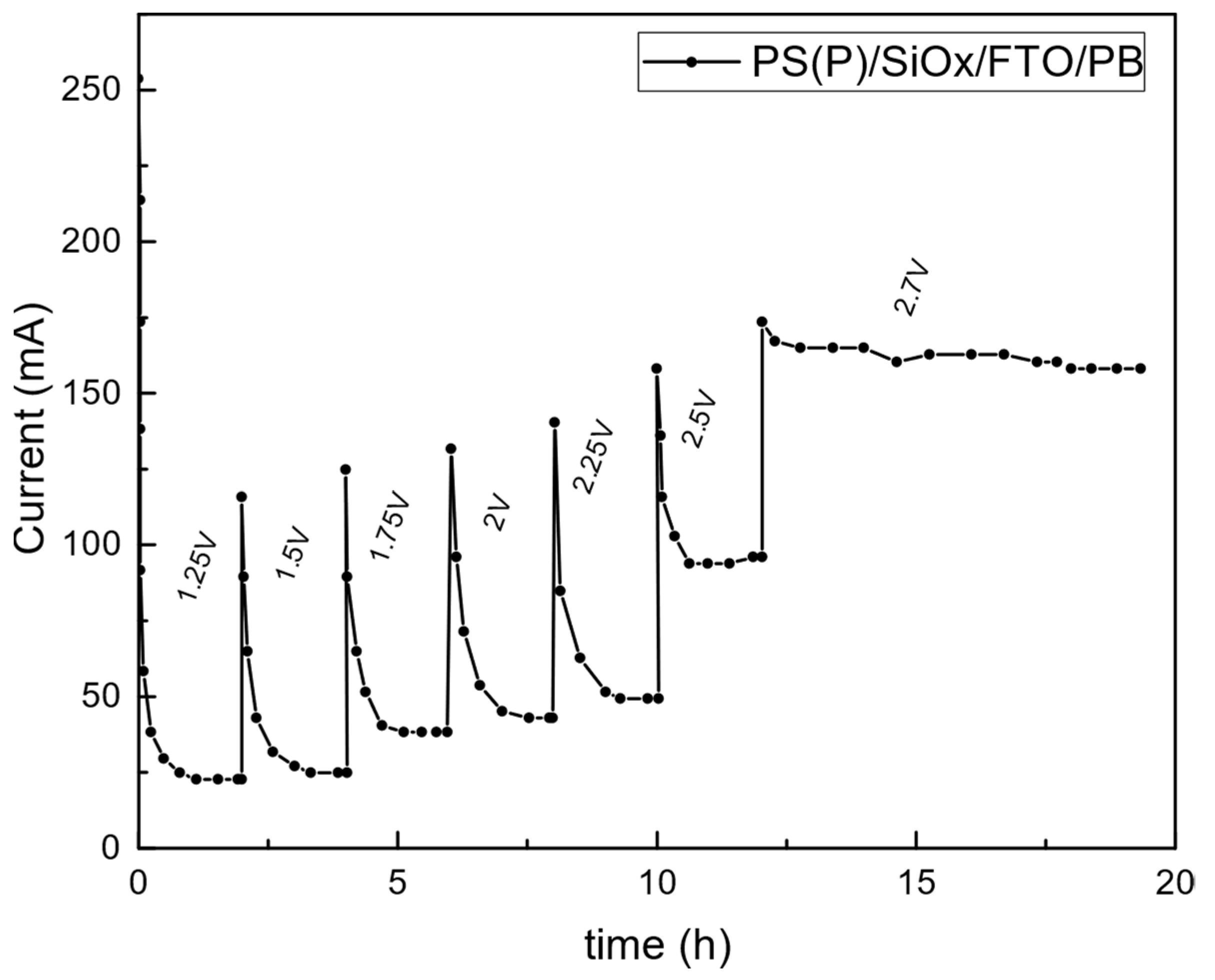
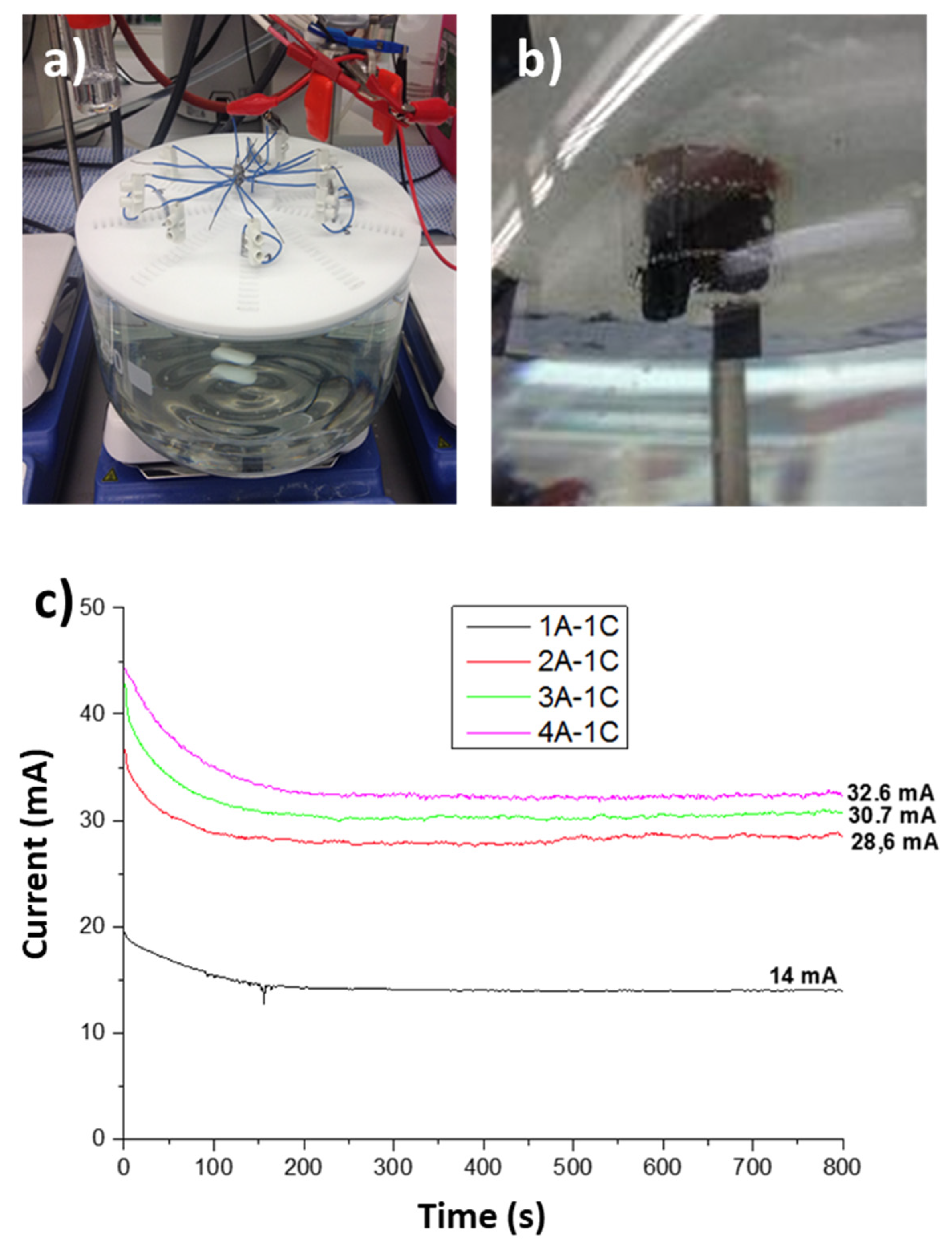
3.3. Prototype Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- El-Dessouky, H.T.; Ettouney, H.M. Fundamentals of Salt Water Desalination; Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Global Issues. Water. 2018. Available online: https://www.un.org/en/global-issues/water (accessed on 4 December 2021).
- Hausmann, J.N.; Schlögl, R.; Menezes, P.W.; Driess, M. Is direct seawater splitting economically meaningful? Energy Environ. Sci. 2021, 14, 3679–3685. [Google Scholar] [CrossRef]
- Farràs, P.; Strasser, P.; Cowan, A.J. Water electrolysis: Direct from the sea or not to be? Joule 2021, 5, 1921–1923. [Google Scholar] [CrossRef]
- O’Brien, T.F.; Bommaraju, T.V.; Hine, F. Handbook of Chlor-Alkali Technology; Springer: New York, NY, USA, 2005; Volume 1. [Google Scholar]
- Bennett, J. Electrodes for generation of hydrogen and oxygen from seawater. Int. J. Hydrog. Energy 1980, 5, 401–408. [Google Scholar] [CrossRef]
- Kato, Z.; Sato, M.; Sasaki, Y.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Electrochemical characterization of degradation of oxygen evolution anode for seawater electrolysis. Electrochim. Acta 2014, 116, 152–157. [Google Scholar] [CrossRef]
- Surendranath, Y.; Dincǎ, M.; Nocera, D.G. Electrolyte-Dependent Electrosynthesis and Activity of Cobalt-Based Water Oxidation Catalysts. J. Am. Chem. Soc. 2009, 131, 2615–2620. [Google Scholar] [CrossRef]
- Dionigi, F.; Reier, T.; Pawolek, Z.; Gliech, M.; Strasser, P. Design Criteria, Operating Conditions, and Nickel–Iron Hydroxide Catalyst Materials for Selective Seawater Electrolysis. ChemSusChem 2016, 9, 962–972. [Google Scholar]
- Ros, C.; Murcia-López, S.; Garcia, X.; Rosado, M.; Arbiol, J.; Llorca, J.; Morante, J.R. Facing seawater splitting challenges by regeneration with Ni–Mo–Fe bifunctional electrocatalyst for hydrogen and oxygen evolution. ChemSuschem 2021, 14, 2872–2881. [Google Scholar] [CrossRef]
- Hsu, S.; Miao, J.; Zhang, L.; Gao, J.; Wang, H.; Tao, H.; Hung, S.; Vasileff, A.; Qiao, S.Z.; Liu, B. An earth-abundant catalyst-based seawater photoelectrolysis system with 7.9% solar-to-hydrogen efficiency. Adv. Mater. 2018, 30, 1707261. [Google Scholar] [CrossRef]
- Hegner, F.S.; Garcés-Pineda, F.A.; González-Cobos, J.; Rodríguez-García, B.; Torrens, M.; Palomares, E.; López, N.; Galan-Mascaros, J.R. Understanding the catalytic selectivity of cobalt hexacyanoferrate toward oxygen evolution in seawater electrolysis. ACS Catal. 2021, 11, 13140–13148. [Google Scholar] [CrossRef]
- Pintado, S.; Goberna-Ferrón, S.; Escudero-Adán, E.C.; Galan-Mascaros, J.R. Fast and Persistent Electrocatalytic Water Oxidation by Co–Fe Prussian Blue Coordination Polymers. J. Am. Chem. Soc. 2013, 135, 13270–13273. [Google Scholar] [CrossRef]
- Alsaç, P.; Ülker, E.; Nune, S.V.K.; Dede, Y.; Karadas, F. Tuning the Electronic Properties of Prussian Blue Analogues for Efficient Water Oxidation Electrocatalysis: Experimental and Computational Studies. Chem. Eur. J. 2018, 24, 4856–4863. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, P.; Reyes-Carmona, A.; Rodríguez-García, B.; Torréns, M.; Morante, J.R.; Arbiol, J.; Galan-Mascaros, J.R. Enhanced activity and acid pH stability of Prussian blue-type oxygen evolution electrocatalysts processed by chemical etching. J. Am. Chem. Soc. 2016, 138, 16037–16045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, V.; Van, M.N.; Lee, H.I.; Kim, J.M.; Kim, Y.; Kim, S.J. A new route for obtaining Prussian blue nanoparticles. Mater. Chem. Phys. 2008, 107, 6–8. [Google Scholar] [CrossRef]
- Goberna-Ferrón, S.; Hernandez, W.Y.; Rodríguez-García, B.; Galán-Mascarós, J.R. Light-driven water oxidation with metal hexacyanometallate heterogeneous catalysts. ACS Catal. 2014, 4, 1637–1641. [Google Scholar] [CrossRef]
- Lehmann, V.; Gösele, U. Porous silicon formation: A quantum wire effect. Appl. Phys. Lett. 1991, 58, 856–858. [Google Scholar] [CrossRef]
- Garcés-Pineda, F.A.; González-Cobos, J.; Torrens, M.; Galan-Mascarós, J.R. Fluorine-doped tin oxide/alumina as long-term robust conducting support for earth-abundant water oxidation electrocatalysts. ChemElectroChem 2019, 6, 2282–2289. [Google Scholar] [CrossRef] [Green Version]
- Garcés, F.; Budini, N.; Arce, R.; Schmidt, J. Effect of thickness on structural and electrical properties of Al-doped ZnO films. Thin Solid Film. 2015, 574, 162–168. [Google Scholar] [CrossRef]
- OSHA. Method ID-126SGX, Chlorine and Chlorine Dioxide in Workplace Atmosphere. 2007. Available online: http://niosh.dnacih.com/nioshdbs/oshameth/t-id126sgx-pv-01-0112-m/t-id126sgx-pv-01-0112-m.html (accessed on 4 December 2021).
- Cheng, F.; Feng, X.; Chen, X.; Lin, W.; Rong, J.; Yang, W. Synergistic action of Co-Fe layered double hydroxide electrocatalyst and multiple ions of sea salt for efficient seawater oxidation at near-neutral pH. Electrochim. Acta 2017, 251, 336–343. [Google Scholar] [CrossRef]
- Izumiya, K.; Akiyama, E.; Habazaki, H.; Kumagai, N.; Kawashima, A.; Hashimoto, K. Anodically deposited manganese oxide and manganese-tungsten oxide electrodes for oxygen evolution from seawater. Electrochim. Acta 1998, 43, 3303–3312. [Google Scholar] [CrossRef]
- Cotillas, S.; Llanos, J.; Rodrigo, M.A.; Cañizares, P. Use of carbon felt cathodes for the electrochemical reclamation of urban treated wastewaters. Appl. Catal. B Environ. 2015, 162, 252–259. [Google Scholar] [CrossRef]
- Biswal, M.; Deshpande, A.; Kelkar, S.; Ogale, S. Water electrolysis with a conducting carbon cloth: Subthreshold hydrogen generation and superthreshold carbon quantum dot formation. ChemSusChem 2014, 7, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Kim, Y.-J.; Kim, J.-H.; Park, M.-S. The effects of surface modification on carbon felt electrodes for use in vanadium redox flow batteries. Mat. Chem. Phys. 2011, 131, 547–553. [Google Scholar] [CrossRef]
- Tong, W.; Forster, M.; Dionigi, F.; Dresp, S.; Erami, R.S.; Strasser, P.; Cowan, A.J.; Farràs, P. Electrolysis of low-grade and saline surface water. Nat. Energy 2020, 5, 367–377. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Cobos, J.; Rodríguez-García, B.; Torréns, M.; Alonso-Almirall, Ò.; Aliaguilla, M.; Galí, D.; Gutiérrez-Tauste, D.; Galindo-Anguera, M.; Garcés-Pineda, F.A.; Galán-Mascarós, J.R. An Autonomous Device for Solar Hydrogen Production from Sea Water. Water 2022, 14, 453. https://doi.org/10.3390/w14030453
González-Cobos J, Rodríguez-García B, Torréns M, Alonso-Almirall Ò, Aliaguilla M, Galí D, Gutiérrez-Tauste D, Galindo-Anguera M, Garcés-Pineda FA, Galán-Mascarós JR. An Autonomous Device for Solar Hydrogen Production from Sea Water. Water. 2022; 14(3):453. https://doi.org/10.3390/w14030453
Chicago/Turabian StyleGonzález-Cobos, Jesús, Bárbara Rodríguez-García, Mabel Torréns, Òscar Alonso-Almirall, Martí Aliaguilla, David Galí, David Gutiérrez-Tauste, Magí Galindo-Anguera, Felipe A. Garcés-Pineda, and José Ramón Galán-Mascarós. 2022. "An Autonomous Device for Solar Hydrogen Production from Sea Water" Water 14, no. 3: 453. https://doi.org/10.3390/w14030453
APA StyleGonzález-Cobos, J., Rodríguez-García, B., Torréns, M., Alonso-Almirall, Ò., Aliaguilla, M., Galí, D., Gutiérrez-Tauste, D., Galindo-Anguera, M., Garcés-Pineda, F. A., & Galán-Mascarós, J. R. (2022). An Autonomous Device for Solar Hydrogen Production from Sea Water. Water, 14(3), 453. https://doi.org/10.3390/w14030453