
Soil Microbiome Study Based on DNA Extraction: A Review

Department of Chemistry, Biology and Biotechnology, Faculty of Civil Engineering and Environmental Sciences, Bialystok University of Technology, 15-351 Bialystok, Poland
Water 2022, 14(24), 3999; https://doi.org/10.3390/w14243999
Submission received: 30 October 2022
/
Revised: 1 December 2022
/
Accepted: 7 December 2022
/
Published: 8 December 2022
(This article belongs to the Special Issue Presence of Microorganisms in Soil and Water)
Abstract
:In recent years, many different methods that allow for the analysis of the biodiversity and structure of the community of microorganisms inhabiting the soil environment have emerged. Many of these approaches are based on molecular methods including the study of genetic biodiversity based on DNA and RNA analysis. They are superior to conventional methods because they do not rely on time-consuming laboratory in vitro cultures and biochemical analyses. Moreover, methods based on the analysis of genetic material are characterized by high sensitivity and repeatability. The abovementioned issues are the subject of many reviews. The novelty of this article is the summarization of the main aspects of soil biological research including genetic techniques, bioinformatics and statistical tools. This approach could be an introduction for scientists starting their work in the field of genetic soil analysis. Additionally, examples of the application of molecular methods in soil research are presented.
1. Introduction
Soil microorganisms play an important role in the decomposition and circulation of organic matter, nutrients or xenobiotics. They are responsible for plant health and nutrition and have an impact on the structure and fertility of the soil [1,2]. Soil is a complex biological ecosystem rich in microorganisms. In turn, according to Singh et al. [3], microorganisms are a very diversified group of organisms and constitute about 60% of the Earth’s biomass, of which the soil environment is inhabited by about 4–5 × 1030 microbial cells. This huge number of microorganisms makes it an unexplored reservoir of genetic diversity. According to the literature data, it is estimated that 1 g of soil is inhabited by about 107 to 109 microorganisms, including bacteria and fungi [3,4,5].
Understanding the need to investigate the genetic diversity of soil microbiota is a challenge for today’s science and an issue of high priority. This is mainly due to the increasing soil pollution resulting from anthropogenic sources and global climate changes as well as other biotic and abiotic factors activity that may modify the composition of the microflora [6]. The traditional cultivation of microorganisms in laboratory conditions by plate method allows for the determination of less than 1% from the total number of microorganisms living on soil. Moreover, the vast majority of microorganisms present in the soil could not be grown in a laboratory. This is due to the fact that most soil microorganisms do not have sufficient growth conditions on artificial culture media. To overcome the challenges, advanced and more comprehensive molecular tools based on the analysis of nucleic acids (DNA and RNA) extracted directly from the soil are now used to assess the biodiversity of the soil environment, without the need to cultivate microorganisms first, which speeds up research procedures [7,8]. The superiority of genetic methods over conventional methods is related to the fact that they do not require in vitro culture, and the targets of the analysis are nucleic acids present in cells. The use of genetic material in the study of soil ecology provides the basis for examining not only selected genes but also complete genomes. The abovementioned approach has revolutionized the concept of determining soil microbial and ecological diversity, and it is currently the main research tool of molecular biology.
The key point in determining soil microbial diversity is the extraction of soil DNA in an appropriate quantity and purity. Contrary to appearances, DNA isolation is a difficult procedure due to the fact that soil is a complex matrix containing, among others, humic acids and various impurities [9]. Humic acids deserve special attention, as they can co-precipitate and inhibit the DNA isolation procedure, and in further steps, lead to the failure of the PCR reaction [10]. Currently, there are many ready-made kits dedicated especially to soil DNA based on extraction on columns or using the magnetic bead system. The most important aspects related to DNA extraction are its quantity and purity, which determine its use for further analysis/manipulation [11].
The use of molecular methods consisting of partial or complete determination of the community of soil microorganisms are powerful and universal tools for assessing the ecology of microorganisms living in their natural environment, thus enabling the connection of ecological processes in the environment with specific populations of microorganisms [12]. Most genetic studies are based on the sequence analysis of the 16S rRNA (prokaryotes) and 18S rRNA (eukaryotes) genes [13]. In addition, molecular methods for biodiversity analysis can be analyzed by type of nucleic acid extracted (DNA or RNA) or by using an analytical approach based on partial or total community analysis. Historical, but still applicable, approaches for the determination of soil microbial diversity have been based on partial (indirect) communities’ studies include, i.e., fingerprints method (e.g., electrophoresis in gradient denaturing gels DGGE, terminal restriction fragments length polymorphism—t-RFLP and single-strand conformation polymorphism—SSCP). Currently, sequencing techniques are more attractive methods for the analysis of environmental biology allowing for a complete analysis of the structure of microorganisms include metabarcoding and metagenomics, which represent high-throughput sequencing techniques such as next generation sequencing (NGS) and third generation sequencing (TGS) [14,15,16].
In the study of the genetic structure of soil microflora, indicators showing the diversity of the community within the sample, referred as α-diversity and between samples (β-diversity), are being used. α-diversity indices include species and phylogenetic richness/evenness/dominance estimators, while β-diversity allows for the assessment of similarity and distance in the community composition [17,18].
The results obtained as part of the genetic tests, especially in NGS, are often difficult to interpret or compare between the studied samples. In order to analyze these results bioinformatics and statistical tools are extremely helpful and even indispensable. Bioinformatics is the set of computer tools used to generate conclusions from molecular biology databases to be able to extract common elements as well as to produce useful predictions. Moreover, bioinformatics tools make it is possible to pre-filter data obtained from genome sequencing, generate gene catalogs and search databases to identify taxa. In turn, defining the interactions of the microbial community requires the use of multidimensional statistical tools [19]. Among the statistical tools, the most commonly used to visualization of statistical results are: principal component analysis (PCA), one-way analysis of similarity (ANOSIM), redundancy analysis (RDA) or principal coordinate analyses (PCoA), non-metric multi-dimensional scaling (NMDS), canonical analyzes of principle components (CAP) and cluster analysis [20,21,22,23].
Although a lot of papers on genetic soil biology issues have appeared in the last 10 years, there is still no available manuscript containing the compilation of almost all methods—from basic to the most advanced, state-of-the-art techniques and tools [16,24,25,26,27,28,29,30,31]. However, it is worth to underline that this paper summarizes basic aspects of the soil molecular biology, and it may provide a good introduction to more detailed content making it useful for scientists starting genetic research in soil microbiome study.
Therefore, the purpose of this work was the review of the most commonly used methods based on DNA investigation currently applied for genetic evaluation of soil biodiversity and taking into account the division according to the scope of community analysis (partial or whole approach) and examples of application. Moreover, the paper also presents examples of bioinformatics and statistical tools facilitating the analysis, interpretation and comparison of research results obtained as an effect of molecular tests.
2. Extraction DNA
2.1. Methods of DNA Extraction
Biodiversity analysis using advanced sequencing technologies focuses on the quantity and, above all, the purity of soil DNA. For this purpose, it is crucial to choose the appropriate DNA extraction method. The soil is characterized by a complex matrix, which may include various inhibitors that may exclude the recovered DNA from further analysis or manipulation. [9]. Humic acids deserve special attention, as their presence may be disturbed by detection and measurement of DNA [10,32].
According to literature data, two approaches for extracting soil DNA are being used in order to determine the genetic diversity of soil microflora. The first strategy involves the indirect isolation of genetic material by extracting cells from a soil sample (about 60 g) and then lysing them [10]. This method allows the separation of bacterial and archaeal cells from eukaryotic cells. The indirect method can be helpful when eukaryotic cells are omitted and when large DNA fragments are analyzed (e.g., to construct fosmids and cosmids clones). The disadvantages of this approach are: the extraction of high amounts of DNA, the inability to analyze eukaryotic sequences and to study interactions between eukaryotes and bacteria/archaeons [10,33]. The second approach is the direct isolation of DNA from a soil sample (e.g., 250 mg) in which the soil matrix is directly mixed with the extraction buffer and the cells present in the sample are subjected to various lysis methods [34]. The genetic material (DNA) released into the solution is then separated from the soil particles and then collected [33]. The advantages of this approach are: high DNA extraction efficiency, high DNA yield and the ability to test various groups of microorganisms. Moreover, due to the fewer steps there is lower risk of the sample contamination [35]. In both the indirect and direct DNA extraction strategies, lysis efficiency and the purification of the sample, mainly of humic acids, are of key importance in determining the soil microbiome. Delmont et al. [10] compared the efficiency of DNA extraction using the indirect and direct method by analyzing data obtained as a result of pyrosequencing. It has been shown that the indirect approach requires an appropriate amount of starting material and is more time-consuming as compared to the direct method. Moreover, as indicated by the literature data, direct methods are most often used in the analysis of the soil microbiome.
Among the manual methods based on direct extraction of soil DNA, various techniques are being used, including with the use of Polyethylene Glycol (PEG)/NaCl, modified mannitol-based methods [36]. CTAB buffer method (Zhou et al., 1996) and many others [37]. Lysis as the first and one of the most important steps in DNA extraction is also different and it can be based on physical lysis (use of beads, repeated freeze–thaw cycles, use of microwaves and ultra-sounds, grinding, digestion with liquid nitrogen), chemical lysis (use of NaCl, SDS, phenol) or enzymatic lysis (using lysozyme or proteinase K). Combinations of these methods are also being used [38]. As reported by Kuske et al. [39] methods based on the use of ultrasound, or cell disruption in an impact mill, result in a significant fragmentation of DNA (5–10 kb). On the other hand, the research of Krsek and Wellington [40] have shown that DNA extracted with beads or by lysozyme and SDS lysis allows for obtaining larger DNA fragments (20–40 kb). The authors also point out that in order to obtain larger DNA fragments (~80 kp), beat-beating and vigorous shaking should be abandoned. Islam et al. [41] compared the different types of lysis used in DNA isolation. It was observed that chemical-enzymatic-mechanical lysis using SDS, NaCl, lysosyme and heat shock was the best in terms of yield and purity of DNA as compared to chemical-enzymatic and chemical lysis. However, according to Felczykowska et al. [38] the choice of the lysis method should depend on the target group of microorganisms (bacteria, fungi, protozoa) due to the different structure of the cell. In addition, the DNA extraction method in the study of the diversity of microorganisms (e.g., in metarbacoding) should be effective for the entire spectrum of soil microorganisms and should not physically disturb the structure of DNA. Studies on DNA extraction efficiency by different methods are summarized in Table 1.
Currently, commercially available DNA isolation kits are most commonly used. The main criteria for selecting a kit are primarily: soil sample size, starting material (soil, sediment, compost, manure), further use of DNA (PCR, qPCR, NGS), laboratory equipment and price. One of the most commonly used soil DNA extraction kit is PowerSoil® DNA Isolation Kit (MOBIO Lab Inc., Carlsbad, CA, USA), which can be used with most soil samples, but studies show that lower isolation efficiency is achieved with samples containing heavy metals or higher amounts of clay and organic substances. In the case of problematic soils, kits taking into account different types of soil are more efficient (e.g., NucleoSpin® Soil kit) [35]. In investigation presented by Knaught et al. [37], it was shown that a greater amount of DNA was obtained after extraction with the NucleoSpin® Soil kit as compared to FastDNA® SPIN kit for soil. Despite the differences in the quantity and quality of the obtained DNA, both tested kits showed a high level of similarity of the genetic profiles obtained by the DGGE technique for the amplified 16S rRNA genes of methanogenic archaea and bacteria. Santos et al. [50] compared three DNA extraction methods in a study of soil protist community. They stated that DNA extracted using the ISO and GnS-GII methods was characterized by low contamination with humic acids, proteins and organic pollutants (e.g., phenol, EDTA). The choice of extraction method may also be important when organic substrates are applied to soils, as reported by Leite et al. [54]. The kits available on the market are mainly based on the direct lysis protocol with the mechanical bead-beating procedure [33]. Kits based on different bead sizes (e.g., PowerSoil® DNA Isolation kit), containing glass beads (E.Z.N.A.® Soil DNA Kit) or ceramic beads (NucleoSpin® Soil kit) are commercially available [35]. However, as indicated by Bürgmann et al. [55] the efficiency of DNA extraction depends on the bead material, speed, time and temperature of disintegration. and mechanical lysis parameters that take into account the use of beads should be selected individually depending on the experiment. Kits in which the DNA binding technology is based on the use of magnetic beads have also appeared on sale (https://norgenbiotek.com/product/soil-dna-isolation-kit-magnetic-bead-system (accessed on 27 November 2022); https://www.omegabiotek.com/product/dna-water-soil-mag-bind-environmental-dna/ (accessed on 27 November 2022)). In this case, it is necessary to have a special magnetic stand. The use of magnetic beads allows obtaining DNA of comparable purity and a similar composition of the microbial community determined by the NGS technique as compared to the classic beat-beating procedure. Moreover, the paramagnetic particle method resulted in a higher DNA concentration with a smaller input sample and was less time consuming [56]. Some kits also offer the possibility of automating the soil DNA isolation procedure using a robot (e.g., Qiagen). An automated or semi-automatic procedure for recovering soil DNA saves time and produces high-quality DNA but may involve higher reagent consumption. There are also commercially available DNA isolation kits in the form of 96-well plates characterized by high throughput [35]. The most commonly used DNA kits together with the manufacturer’s recommended sample size, material type and downstream application of DNA are shown in Figure 1.
In order to meet the high requirements that the DNA used to study the soil metagenome must achieve, commercial methods are often further modified to obtain DNA of the highest possible quantity and/or quality and representative genetic diversity [11]. Protocols are more or less modified and improved depending on organic matter and colloid content, as well as components that are potential inhibitors (e.g., metal ions). Modifications and improvements are used primarily at the DNA lysis and purification stage as additional steps to commercial protocols. Some manufacturers propose the use of alternating freezing (at −70 °C) and thawing combined with beat-beating, but the number of such manipulations should be limited to a maximum of three times (https://eurx.com.pl/docs/manuals/en/e3570.pdf (accessed on 27 November 2022)). For problematic samples containing multiple potential PCR inhibitors to downstream NGS, extraction based on beat-beating with heat exposure is recommended [56]. In study by Adhikari et al. [57] soil sample intended for DNA extraction was mixed with a sterile buffer containing gelatin and after 10 min of incubation, the soil solution was used in a commercial protocol. In conclusion, the choice of an appropriate method/kit for soil DNA extraction should be dictated by the lysis efficiency, the amount of extracted DNA, the efficiency of the recovery of the richness of microorganisms and the purity of DNA [11].
2.2. The Main Factors Influencing on Yield of DNA Extraction and Its Downstream of Application
According to Plassart et al. [51] and Wüst et al. [58], the efficiency of DNA extraction from soil depends on the type of soil and its properties. The content of organic matter, clay, silt, water and pH are the main factors determining the quality and quantity of the obtained DNA. These factors determine the growth of certain microorganisms, the formation of aggregates that are the habitat of microorganisms or the binding of DNA with some soil components. According to Plassart et al. [51], it turned out that a greater amount of DNA was extracted from calcareous arable soil as compared to sandy forest soil with a high organic carbon content and a high C: N ratio. However, these parameters may also influence the purity of the isolated DNA and, subsequently, the PCR efficiency, as reported by Sagova-Mareckova et al. [59].
Another factor that determines the quality and quantity of the isolated DNA is the type of soil microorganisms or the target DNA under investigation. The lysis stage is of decisive importance here, the success of which depends on the structure of microorganisms, including the number of Gram-positive and Gram-negative bacteria. [32]. However, the studies demonstrated by Santos et al. [60] indicate that the choice of DNA isolation method is important in determining the diversity of the protist community. Often, commercially available protocols need to be optimized for DNA isolation to determine the diversity of target microorganisms.
According to Morita et al. [61], the purpose of the isolated DNA depends on the size of the sample, which is important in the PCR-DGGE analysis as well as in the NGS approach. The authors observed differences in microbial community structure between two sizes of soil samples (0.2 g and 1 g) in different taxonomic ranks (order, class, and phylum). In addition, they concluded that the 0.2 g soil mass was preferable due to possible higher shearing force for DNA extraction and lower costs associated with the consumption of reagents and other consumables. In addition, the sample size for DNA extraction may be indirectly related to the properties of the soil. For example, the consortia of bacteria and fungi are sensitive to changes in pH. At a pH of around 7, bacteria are likely to increase, while fungi will remain at a similar level or decrease at a higher pH [62,63]. Therefore, it may be necessary to increase the weight of the sample in order to obtain repeatable and comparable results. For sequencing techniques, a sample weight of 0.2 to 0.5 g is currently acceptable and optimal for most research purposes [16]. In contrast, the studies presented by Leite et al. [54] indicate that the extraction of DNA from soil with the addition of biochar gives different numbers of profiles in the PCR-DGGE analysis after using different extraction methods. The key here is the presence of biochar, which has a strong adsorption to the phosphate backbone of DNA. In addition, it is a substrate rich in organic matter, soluble ions such as ammonium, nitrate, phosphate, which are difficult to remove and may inhibit PCR. In addition, the efficiency of DNA recovery may be more difficult in contaminated soils because contaminants can inhibit enzyme activity during lysis and reduce PCR efficiency. Mazziotti et al. [11] demonstrated that the choice of DNA extraction method is relevant in soils contaminated with benzo(a)pyrene. Therefore, it is recommended to test several DNA extraction methods before starting the experiment in order to optimize and obtain reproducible results.
The quality and quantity of DNA used in metagenomic studies is also influenced by the transport and storage time of samples. The storage time of samples for metagenomic studies should be as short as possible, and the analyses should be performed as soon as possible to prevent contamination of the sample and/or its destruction, which will impact on the reliability of the results [38]. If this is not possible, samples should be frozen or stored in commercially available protein inhibitors [35]. The study by Guerrieri et al. [64] revealed that the method of soil sample preservation influenced DNA metabarcoding results, especially for rare taxa. In addition, the researchers proposed guidelines for the selection of an optimal soil sample preservation conditions for metabarcoding.
The choice of method for extracting soil DNA should also be based on the further purpose of the DNA, including the technique for studying genetic diversity. For example, Stach et al. [65] observed that different methods of direct DNA isolation may provide different results in PCR-SSCP analysis (PCR–single strand conformation polymorphism). In addition, they highlighted that higher DNA yields do not correspond to higher sequence diversity. Soil DNA extraction methods may impact both the phylotype abundance and composition of the indigenous bacterial community identified by amplified ribosomal DNA restriction analysis (ARDRA) and ribosomal intergenic spacer analysis (RISA), as reported by Martin-Laurent et al. [47]. In addition, Feinstein et al. [66], using qPCR, T-RFLP and pyrosequencing individual soil samples, suggested that the error resulting from DNA extraction may be negated for some analyses by combining three subsequent extractions representing the majority of DNA obtained in a multiple extraction procedure. Zielińska et al. [67] proved that the extraction method can affect not only the quantity and quality of DNA, but also the structure of microbial communities obtained by the NGS approach. The authors also indicated that in this type of study, genetic diversity can also depend on the sequencing technology and bioinformatics tools used. The research conducted by Basim et al. [52] determined the applicability of four different DNA extraction protocols for sequencing and identifying native bacterial species involved in the remediation of oily soils. The results showed that the highest yield and quality of DNA was obtained using a protocol that included extraction with silica beads, triple freeze–thaw and chemical lysis with the use of lysozyme. Sakai [33] compared various methods for extracting high-molecular-weight (HMW) DNA from soil, which is useful for studying the function and diversity of soil microorganisms. Manipulations such as digestion of the sample in liquid nitrogen, treatment for 1h with lysozyme at 45 °C, and incubation at 50 °C with protease and SDS for 5 h increased the quantity and efficiency of HMW DNA extraction. In addition, for some soils (e.g., Andosols) the addition of boiled sonicated salmon DNA may increase HMW DNA extraction efficiency. Kaden et al. [4] reports that the combination of several different DNA extraction methods allows the detection of a large number of bacterial species in soils with natural nanoparticles. They also state that the amount of DNA isolated does not correlate with the number of species detectable
3. Soil DNA-Based Methods
3.1. Partial Analysis of Soil Microbiota
In recent decades, techniques known as “fingerprints”, such as DGGE, T-RFLP, and fluorescent hybridization in situ, have gained popularity in studying the organization of the microbial community [68]. It is a set of molecular biology methods that can be used to determine the diversity profile of a microbial community quickly and relatively easily. In addition, the fingerprint methods make it possible to analyze multiple samples simultaneously allowing for comparison of genetic diversity from different habitats or over time. Rather than directly identifying or counting individual cells in an environmental sample, these techniques show how many gene variants are present. It is generally assumed that each other gene variant represents a different type of microorganism. The results obtained as a result of these methods show a certain pattern (profile) characteristic for a given sample/habitat [69].
Genetic fingerprinting techniques are based on the isolation of soil genomic DNA, which is then purified and subjected to PCR amplification. The most frequently amplified are ribosomal RNA (rRNA) genes as a molecular marker, and they mainly include: 16S rRNA (bacteria and archea), 18S rRNA and internal transcribed spacer (ITS) regions (fungi) [24,70]. In general fingerprinting techniques are a fast, reproducible, reliable and relatively inexpensive. For example, the T-RFLP approach produces comparable genetic profiles as next-generation sequencing (via 454 and the Illumina and Ion Torrent platform) [68]. The key point is that simple fingerprinting methods can still capture important changes in the population that are critical to soil environmental quality control [71]. The basic drawbacks associated with the fingerprints approach are mainly problems related to PCR optimization (selection and design of primers; specificity of the PCR condition), restriction enzyme selection (T-RFLP approach), limited phylogenetic identification associated with the fact that they band can represent many species or one species can give many bands (DGGE, T-RFLP). Moreover, in some methods only small fragments/sequences can be analyzed (DGGE, SSCP) [69,72,73]. Considering T-TFLP assay, currently the analysis of obtained genetic patterns is also possible in silico (e.g., based on fragments size profile: TRIFLE). Gao et al. [74] used T-RFLP and DGGE techniques to determine the effect of the biocontrol agent Pseudomonas fluorescens 2P24 on the fungal community in the cucumber rhizosphere. The results obtained by the T-RFLP approach differed depending on the restriction enzyme used, while the data obtained by the DGGE method showed an initial shock to the fungal structure associated with the presence of P. fluorescens, which recovered as the amount of biocontrol agent decreased. Smalla et al. [6] compared DGGE, T-RFLP and SSCP assays using the 16S rRNA gene amplicon. The results showed that the T-RFLP approach appears more useful for routine analysis, because it does not require comparison between gels. In contrast, the DGGE and SSCP methods allow easier characterization of differentially expressed bands by cloning and sequencing. However, the patterns obtained by the methods tested showed similarity in bacterial community composition and correlated with the properties of the soils analyzed. Wang et al. [75] examined the effect of metalaxyl on the community of soil microorganisms (bacteria and fungi) using DGGE and T-RFLP approaches. Both methods revealed the no effect of this pesticide on the microbial community. The T-RFLP method allowed examining the community structure by comparing the relative abundances of identified bacterial T-RFs in a sample. In turn, patterns obtained by DGGE profiling were compared using PCA analysis and calculating α-diversity estimators (Shannon–Wiener, Simpson’s and Evenness indices). A research described by De Vrieze [68] compared the microbial community using T-RFLP and Illumina techniques. Using β-diversity analysis a high degree of similarity between the profiles was demonstrated. It was also suggested that the T-RFLP approach is worth applying to monitor changes in overall microbial community structure due to the simplicity and potentially low cost of the analysis. Gryta et al. [76] compared a single T-RFLP method with multiplex T-RFLP for the detection of bacteria, fungi and archaeons. The genetic profiles obtained with the two protocols were similar, especially for bacteria and fungi, which supports the use of the multiplex approach to reduce cost and time analysis.
Analyzes based on DNA amplification are a key tool in the diagnosis or testing of food. They are also applied in the analysis of complex matrices such as environmental samples, including soil. In soil research, they are mainly used to assess the number of microorganisms (archaea, bacteria and fungi), or to diagnose plant pathogens. They are used to detect functional gene markers likewise [12,77]. One of the widely used PCR techniques is real-time PCR or qPCR (quantitative PCR). This technique complements fingerprint and sequencing methods. Quantitative PCR widespread in the area of microbial ecology for the examination of the genes number (or transcripts) present in environmental samples [78]. qPCR is based on the process of an intercalation of a fluorescent dye (most often SYBR Green) or a fluorescent probe (TaqMan probes, 5′-nuclease probes) in order to monitor an increase in the amount of amplified product in real time. It also provides insight into the kinetics of the reaction and therefore allows for the estimation of the amount of product at the beginning of the reaction [12]. The disadvantage of qPCR is the possibility of interference of the fluorescent signal by inhibitors contained in the sample, which may occur when using difficult matrices [15]. Moreover, as reported by Kim et al. [79], qPCR requires the performance of a calibration curve to determine the amount of amplified product, which is often an expensive and labor-intensive process. In addition, the presence of humic compounds present in the soil may interfere with the performance of the standard curve and thus the proper quantification of the PCR product. Moreover, effectiveness of RT-PCR is also affected by reagents, their purity, optimal concentration, selection of polymerase and primers.
A digital PCR droplet (ddPCR) has been introduced as an alternative quantitative PCR technique [79,80]. This method, like qPCR, is based on fluorescently labeled nucleotides, but it is based on a different method of measuring the amount of amplified product. In addition, it uses the technique of water-oil emulsion, thanks to which a single reaction is fractionated into many drops (approx. 20,000). There is an amplification in some drops where the specific primers were occurred. The system then reads the reacted (positive) and all the other (negative) molecules, from which the absolute number of target molecules in the sample is generated, without the need to refer to the standard curves or to the reference gene The disadvantages of ddPCR include the necessity of the dilution of DNA samples, especially for bacteria that are densely populated; it is an expensive, laborious and time-consuming method because it requires the production and reading of the droplet, which involves a greater number of stages of analysis. A study by Liu et al. [81]. Showed that using ddPCR instead of PCR and real-time PCR provided a more accurate and sensitive method for detecting T. controversa teliospores in soil. In contrast, results presented in Voegel et al. [82]. revealed that the ddPCR technique is a valid alternative to qPCR for quantifying genes involved in nitrification or denitrification. Its use in soils is supported by the fact that it is highly sensitive and allows the determination of even small amounts of a target gene. In addition, it has a low susceptibility to high concentrations of PCR inhibitors.
The main division of techniques for studying the diversity and structure of microorganisms, including both direct and indirect techniques, is presented in Figure 2.
3.2. High-Throughput Sequencing Techniques
Currently, the microbial community in soil is defined by using modern molecular biology methods, which are based on the isolation of the total DNA from soil and its sequencing. The analysis can be based on two approaches. The first is related to the identification of the composition of the microorganism community by determining the sequence of barcode marker genes (metabarcoding, target metagenomics). The second approach is related to the determination of the genes collection of the community inhabiting a studied environment (metagenomics, global metagenomics, shotgun metagenomics) [16,25]. It involves the identification of the taxonomic composition of the community from environmental samples by amplifying the sample and then high-throughput sequencing of DNA barcode such as 16S, ITS or 18S. Sequencing techniques based on the amplification of marker genes are fast, cost-effective and well-studied. Possible errors that may arise as a result of them are related to the representativeness of the sample, the affinity of the selected primers to all possible DNA sequences present in the sample, the selection of the appropriate amplification region, the size of the amplicon or the selection of the number of PCR cycles [16,25,83].
The global metagenomic technique gives more detailed results and higher taxonomic resolution as compared to metabarcoding. On its basis, it is possible to determine, inter alia, the relative abundance of functional genes is free from systematic errors related to PCR, is not targeted at specific microorganisms (it captures fungi, bacteria, viruses or others) and can be applied to obtain a novel gene family [25,84]. In turn, DNA metabarcoding techniques are common because of their relative simplicity and relatively low cost. The target metagenomics approach is most often used in the analysis of archaea and bacteria communities, and it is also possible to analyze the structure and diversity of fungi, algae and protozoa. As a result, it is possible to create a list of taxa with their relative abundance, which can then be used in α-diversity and β-diversity analyzes, determine the composition and estimate the shares of indicator taxa in the community. It is also possible to metabarcode functional genes and determine the composition of communities associated with them. However, the amount of functional information is limited as compared to global metagenomics. In addition, the obtained data as a result of barcoding allow to determine the correlation between the identified taxa/communities and the ecological conditions of the soil. It is important that target metagenomics play a leading role in the study of ecology and distribution of poorly studied taxa of soil microorganisms. [16,25,83]. According to Knight et al. [25], in order to obtain a high-level overview but at low resolution, the use of metabarcoding is recommended, while for a detailed analysis of total DNA in a sample at a high-resolution level (species or strain level), the use of global metagenomics techniques is preferable.
Both metabarcoding and metagenomics in soil biology have limits and constraints that pose a constant challenge to scientists creating and optimizing research methodologies. One of the main problems of soil analysis is related primarily to the properties of the soil, which is a complex matrix with variable composition and properties that create problems with the optimal mass of the sample and the representativeness of the test samples. Another problem arises from the presence of inhibitors such as humic acids and other organic substances, and the absorption of microbial cells on soil particles, which reduces the efficiency of DNA extraction and purification. This is of great importance for target metagenomic techniques where DNA extracts contaminated with humic acids can inhibit PCR. A common problem of molecular techniques based on PCR amplification of environmental DNA is the probability of chimera formation. These are sequences of artifacts between multiple parental sequences that may be misinterpreted as a new microorganism, thereby increasing the apparent diversity [85,86]. In this case, it is important to choose the appropriate kit for the soil DNA extraction, optimize PCR and select reagents of high purity. The third problem is related to the large diversity of communities characterized by different physiological states (active, potentially active, dormant and dead) and the presence of extracellular DNA [16,25,72].
Next-generation sequencing (NGS), part of the high-throughput sequencing (HTS) techniques, is now the state-of-the-art technique for fast and efficient genome sequencing. Sequencing of the many molecular markers with this technique allows us to explain the complexity of microbial communities, including those inhabiting the soil. Next-generation sequencing is a non-Sanger high-throughput sequencing technology. Millions of DNA fragments can be sequenced simultaneously (mass parallel sequencing), which increases greatly the efficiency of sequencing. The advantages of this technology, unlike the first-generation techniques of sequencing, include that it is high-throughput, its cost reduction and sequencing duration, as well as its high precision [87]. In NGS sequencing, the genome is usually fragmented into small fragments, which are then randomly sampled and sequenced using the technique of choice. In an automated process, multiple fragments are sequenced simultaneously, which allows for the determination of this “massively parallel sequencing” (MPS) method. The most popular NGS techniques include pyrosequencing, SOLiD, Illumina HiSeq and MiSeq Sequencing [24]. Among the third-generation sequencing platforms Pacific, NextSeq, MinION and BioNano can be distinguished [24,88,89,90].
The pyrosequencing technique was developed by 454 Life Science, acquired by Roche Diagnostics. It was the first commercially available successful next-generation system. Pyrosequencing technology is based on the detection of pyrophosphate released during the incorporation of a nucleotide into a newly synthesized DNA strand [91]. Released pyrophosphate is measured by two sulfurase and luciferase enzyme reactions. As a result, a stream of photons is created, recorded by a camera with a photosensitive CCD matrix (Charche Coupled Device). The genomic DNA fragments are ligated to short fragments of a given sequence (adapters) as a template for the PCR and amplified on synthetic beads in a water-in-oil emulsion (emulsion PCR). During the PCR reaction, the beads bind the replicated DNA molecule. The bead-amplified DNA templates are then placed into the wells of a 44 µm diameter microplate containing the appropriate enzyme cocktail, such as that one copy of the DNA fragment corresponds to one bead and one well. These steps replace laborious cloning and avoid errors caused by it. The loaded slide is then loaded into a sequencing machine that washes deoxynucleotides over the plate, extending the DNA template chains in each well and promoting photon release. The computer records the light release, records the DNA sequence in each well, and eventually interprets this data to align smaller sequence fragments with the complete genome sequence [91,92,93]. The advantages of pyrosequencing include possibility of sequencing samples taken from multiple environments in a single run, while after the sequencing process the readings can be analyzed by their assigned nucleotide barcode which is added to the templates during PCR. In addition, it allows you to generate up to a million readings and read sequences of up to 700 bp on the FLX sequencer [12,24,91,94].
Currently, the microbiological diversity of the soil environment based on the extracted DNA is most extensively analyzed using Illumina sequencing on the MiSeq and HiSeq platforms which were launched in 2006 [24,26]. In Illumina technology, sequencing is based on synthesis, which generates more data per run and thus reduces the cost of analysis. In addition, it uses a flow cell with an attached field of fluorescently labeled oligonucleotides. During each sequencing cycle, a single labeled deoxynucleoside triphosphate (dNTP) is added to the nucleic acid chain. The terminator for the polymerization is the nucleotide label. For base identification, a fluorescent label is imaged after each dNTP attachment. Attachment of another nucleotide is possible because of an enzymatic digestion. Nucleotide insertion errors are minimized by the presence as separate single molecules of the four reversible terminator-bound dNTPs (A, C, T, G). The end result is very accurate base-by-base sequencing. The advantages of using the Illumina platform include, above all, the breadth of application, simplicity, high throughput and high efficiency, short operating times and the possibility of scaling. The disadvantages include the fact that it is characterized by a very small number of all reads, moreover, the cost of purchasing the instrument is very high [24,95].
The Ion Torrent sequencing technology is an example of another NGS platform. This NGS approach is based on semiconductor chips. Pre-treatment of template in Ion Torrent occurs using the emulsion PCR. PCR components, particles surrounded by primers and low concentration template fragments are combined with the oil to generate microreactions (picolitre). Then, the particles are then subjected to thermal cycles in which the clonal amplification of individual DNA molecules on the surfaces of individual particles takes place. Next, the particles are deposited in separate nano-well chambers on a semiconductor sequencing chip. Successive nucleotides are cyclically introduced in the presence of a DNA polymerase, and the successful inclusion of a specific nucleotide results in the release of hydrogen ions, which is recorded [96]. The advantage of using this solution is low price and high accuracy, while the disadvantage is relatively short readings and slow operation [24]. Salipante et al. [96] compared different sequencing platforms (llumina MiSeq and Ion Torrent Personal Genome Machine (PGM)) to characterize the bacterial community by sequencing the 16S rRNA (V1-V2) amplicon. The researchers reported relatively higher error rates with the Ion Torrent platform and observed premature truncation of reads. Read truncation was dependent on the target species and sequencing direction
In 2006, Life Technologies designed the NGS technology based on sequencing by ligation and oligonucleotide detection (SOLiD) [91]. The SOLiD approach uses, in contrast to the below-mentioned method, sequencing by ligation using emulsion PCR with small size magnetic beads to amplify DNA fragments during parallel sequencing. DNA ligation is performed in order to join specific fluorescently labeled 8-mer oligonucleotides for “dinucleotide encoding”, the 4th and 5th bases of which are encoded by specific fluorescence. Each 8 m fluorescent marker identifies a combination of two bases that can be further distinguished using a universal primer offsetting scheme. The primer offsetting scheme refers to a universal primer that is shifted one base from the position of the adapter fragment to hybridize to DNA templates in five cycles allowing the entire fragment to be sequenced and double sequencing at each base position during each cycle. Each ligation step involves fluorescence detection and another round of ligation [97]. The weakness of this technique is that it is time consuming. It is an attractive method because it generates low costs and is characterized by high accuracy [24,97].
The second-generation sequencing application has greatly improved soil biological analysis, but it has still not met some of the difficulties, such as assembling and defining complex genome fragments, detecting methylation, errors during PCR, etc. Many of these limitations have been removed or reduced by the introduction of third generation sequencing (TGS). They are represented by Pacific BioSciences (PacBio) real-time single molecule sequencing (SMRT), nanopore sequencing by Oxford Nanopore Technologies (PromethION and MinION), BioNano Genomics (BioNano) sequencing or NextSeq Sequensing by Illumina. They provide very long length readings (>20 kb) as compared to NGS, and they are applied in order to create highly accurate de novo assemblies composed of hundreds of species of genomes. This solution provides lower cost and shorter time without sacrificing quality of assembly of the genome. An example of the disadvantages of the TGS approach is the need for a huge space for data storage and an enormous computing power (SMRT). High cost per read and the possibility of biased errors (MinIon) should be also mentioned. However, despite the disadvantages, TGS technologies are fast, simple and they are now becoming a very attractive and future-proof solution in the analysis of both amplicon-based sequencing as well as metagenomics of soil [24,89,90].
4. Bioinformatics and Statistical Analysis in Diversity Estimation
4.1. Bioinformatics
An intensive development of molecular methods has led to the collection of a huge amount of experimental data. Both the large amount of obtained results and the data sets resulting from cell-scale experiments cannot be used without the help of advanced computer technology. Nowadays, informatics is an applied science, and computer is now a key part of projects used to determine the sequence, structure, or other type of data. Computer programs used to generate conclusions from molecular biology data archives are able to extract common elements as well as to prepare useful predictions. Additionally, it should be mentioned that the natural environment of computer science is the Internet. To achieve their scientific goals, most projects start by identifying the necessary data on one or more websites. The found data is then analyzed, usually using tools available on the web [97,98].
The results of an experimental research in molecular biology are stored in computer databases. Databases are sets of data stored in a strictly defined manner, corresponding to the type of resources collected, and software used to search them. Modern biological databases resemble popular internet portals. In addition to keyword searches, biological databases also offer specialized methods for searching biological data types, such as sequences, structures, and molecular interaction networks. The assumptions of the algorithms that perform such tasks are based on biochemical knowledge and molecular evolution. DNA sequences are the primary type of data collected in biological databases on DNA testing. All newly learned DNA sequences are deposited in one of three databases: GenBank, EMBL, DDBJ. These databases automatically exchange information so that the sequence deposited in one of them is also in the other two. Research work requires the deposit of the newly learned sequences in one of these databases before the publication of the article [98,99,100,101,102].
Searching of the sequence databases is to find all sequences similar to the newly learned sequence. The solution to of this problem is possible only by analyzing the statistical significance of the sequence similarity coefficient. Undoubtedly, the most commonly used method for searching databases is the BLAST method (now PSI-BLAST), in which statistical significance is calculated as expect value (e-value). The BLAST program offers five variants of database searching: blastp (protein sequences), blastn (DNA and RNA sequences), blastx, tblastn, tblastnx. Another tool designed to match the DNA or protein sequence readings to the protein reference database is DIAMOND [21,99,100].
Bioinformatics tools are also used for phylogenetic analysis aimed at determining the evolutionary history of a group of similar DNA or protein sequences. Phylogenetic compounds are described as trees. The most commonly used tools for creating phylogenetic trees (and not only) include: Phylip or MEGA (Molecular Evolutionary Genetics Analysis) [70]. In addition, the website www.phylogeny.fr offers access to many tools, including aligning multiple sequences, creating phylogenetic trees and viewing them [99].
Among current and the most popular tools for amplicon-based sequencing DADA2, QIIME2, Deblur and CoMa were applied [16,30,102,103,104].
DADA2 is an open source of R-package that is based on DADA (Divisive Amplicon Denoising Algorithm) algorithm and improves it. DADA2 allows full workflow with amplicon, which include filtering, dereplication, sample inference, chimera identification or merging of paired-end reads. The DADA2 approach is an algorithm for the inference of the ASVs [30,104].
QIIME2 (Quantitative Insights Into Microbial Ecology) and Mothur are the pipeline for preparing microbiome analysis from raw sequencing data through visualization, quality control and statistics. The disadvantage of using these solutions is the need to have extensive knowledge and experience in the use of command line tools and data processing based on sequences. Partial solution to these problems is to the possibility to use the Internet blogs of users and numerous online documentation [30,102].
CoMA (Comparative Microbiome Analysis) is a free and intuitive pipeline compatible with any common operating system such as Linux, Windows, macOS. This approach is very flexible usage with a huge variety of tools and options such as quality checking, clustering to operational taxonomic units (OTUs), taxonomic assignment, data post-processing, data visualization, and statistical appraisal. The tool can be applied in soil microbiome studies [102].
Another tool—Deblur—uses error profiles to derive putative error-free sequences from the Illumina MiSeq and HiSeq sequencing platforms. Deblur significantly reduces computational requirements over similar sOTU (sub-operational-taxonomic-unit) methods, unlike DADA2, and it does so with similar or better sensitivity and specificity. It is released under the Berkeley Software Distribution (BSD) open-source license and allows easy commercial deployment. Deblur provides a quick and sensitive way to assess ecological patterns resulting from the differentiation of closely related taxa [94]. Comparing various bioinformatic pipelines for microbial 16S rRNA amplicon sequencing was presented in paper by Prodan et al. [105]. Testing of various pipelines authors demonstrated that DADA showed the best sensitivity and resolution, but was characterized by the generation of more spurious ASVs in comparison with USEARCH-UNOISE3 and QIIME2-Deblur. In contrast, USEARCHUNOISE3 presented the best balance between resolution and specificity. The researchers also noted that QIIME-uclust produced a large number of false OTUs, and overestimated the measures of alpha diversity, suggesting the use of other pipelines. Hupfauf et al. [102] compared different bioinformatics pipelines (CoMA, Mothur, QIIME and QIIME2-DADA2) by analyzing microbial community sequencing data of three different soils. All the compared tools generated similar results and revealed the majority of all genera in the mock communities.
Searching these databases concerns mainly sequences obtained with the NGS technology. Currently, the websites also provide programs for predicting potential microorganism species obtained by fingerprint methods (T-RFLP), such as the phylogenetic assignment tool (PAT), TRUFFLE, APLAUS. They allow for the analysis of the microbial community composition based on the comparison of tRFs predicted from in silico rRNA sequence databases. However, identification using these tools is laborious and does not give reliable results compared to the results obtained with NGS [94]. After sequencing, the obtained sequences are analyzed using a bioinformatics tool. OTUs (operational taxonomic units) assigned, sequence folded, and annotated for phylogenetic characterization are prepared [97]. The clustering OTUs approach is based on the assumption that similar microorganisms will have similar target gene sequences and that rare sequencing errors will have little effect on the consensus sequence for these clusters. Clusters are often generated using a similarity threshold of 97% sequence identity. This approach carries the risk that many similar species can be grouped into one OTU and their individual identifications will be lost to cluster abstraction [106]. As an alternative to OTU, amplicon sequence variants (ASVs) known also as ribosomal sequence variants (RSVs) are applied. This approach is inferred by a de novo process and allows to group and merge sequences based on forward and reverse single nucleotide readings. In other words, this approach allows for the determination of which sequences exactly have been read and how many times each of them has been read. This data will be combined with an error model for the sequencing run, allowing comparison similar readings to determine the probability that a given reading at a given frequency is not due to a sequencer error. According to the ASV assumption, a given target gene sequence should always generate the same ASV, and a given ASV, being the exact sequence, can be compared with the reference database at a much higher resolution, which allows for more accurate identification down to the species level, and even potentially beyond. The main advantage of the ASV approach is the more precise identification of microorganisms as well as it can provide a more detailed picture of the diversity in the sample. Moreover, ASV approaches tend to be less time-consuming than OTUs, and no arbitrary threshold and centroid selections are needed [16,102,107]. For instance, ASVs/RSVs approach was used in the investigation by Semenov et al. [108] on the analysis of the plant rhizosphere microbiome. In some papers, OTUs are also presented for the results obtained with the fingerprinting method (e.g., t-RFLP), however the predicted OTUs do not reflect the specific OTUs obtained with the NGS approach [6,109].
In the case of shotgun metagenomic data analysis, the most popular tools include MG-RAST, MEGAHIT and IMG/M [15,26,27]. MG-RAST is a web-based metagenomics tool using to profile the function and composition of microbial community. MG-RAST provides resolute analysis and visualization of data directly via internet platforms or tools such as matR (metagenomic analysis tools for R). The limitation of the tool can be server overload and other factors specific to working online. It is a platform that is constantly changing to meet the needs of new sequencing technologies and growing data volume [16,30]. Another tool—MEGAHIT, is a NGS de novo tool for assembling large and complex metagenomics data in a time- and cost-efficient manner. Additionally, MEGAHIT enables an efficient assembly of large and complex metagenomics data on a single server, while giving better completeness and contiguity [110]. The Integrated Microbial Genomes & Microbiomes (IMG/M) is a user driven data management resource that enables users worldwide to analyze microbial genomes and metagenomes in a comparative context. IMG allows users to search and view data and to perform many analyzes directly on the platform user’s Interface. Like other web-based interfaces, it is still being expanded and improved to support new data types [111].
4.2. Statistics
In order to improve the analysis and interpretation of the results from the molecular tests and to obtain answers to questions about changes in soil biodiversity as an effect of pollution or various treatments, various indices and indicators showing abundance, richness and variability of soil microflora have been introduced. The OTUs and ASVs/RSV generated as a result of the NGS approach (and OTUs estimated in some fingerprint method, e.g., T-RFLP) are the basis for the calculation of parameters illustrating biodiversity and its changes in soil.
On the basis of the sequencing data, appropriate indicators are calculated, allowing the comparison of the structure of microorganisms both in a sample (α-diversity) and between samples (β-diversity) [17,18]. The division and examples of biodiversity indicators together with an example of visualization are presented in Figure 3.
α-diversity is based on the measurement of species richness which can be expressed as the number of species/OTU, relative abundance, Chao- 1 abundance estimator, richness and evenness estimators (Shannon-Weaver, Margalef or Hill indices), dominance indices (i.e., Gini-Simpson, Berger-Parker indices). In addition, these include measures of phylogenetic diversity, which are sensitive to the number of sequences per sample [25]. These measures can be compared using traditional statistical approaches such as the t-test, parametric analysis of variance (ANOVA), or the nonparametric Kruskal–Wallis test [31]. OTU sequencing results are often analyzed by generating a rarefraction curve that allows standardize unequal sequencing effort and enable comparison of different runs or replicates [112]. As Beisel et al. [113] indicate, if using α- diversity estimators, the evenness measure should be used together with richness and diversity indices, which can greatly facilitate data interpretation and reduce the importance of independence between measures of evenness and richness of microbial communities.
The β-diversity measurements are based on the measurement of (dis) similarity and distance between each pair of samples. Qualitative indicators such as Jaccard index, unweighted UniFrac are measured based on presence/absence of features, while quantitative indicators (Bray–Curtis, weighted UniFrac) use feature abundance measurement [17,18,25].
The OTUs and ASVs/RSV generated by sequencing can be analyzed using various statistical tools designed for multivariate analysis and the visualization of the relationship between the data includes ordination plot or cluster analysis. Their task is to organize, systematize and compare the results, visualize them and find possible links between changes in biodiversity in response to environmental factors. One way analysis of similarity (ANOSIM) is a non-parametric statistical test commonly used in ecology. The test allows to check if there are significant similarities between two or more groups of sampling units. This analysis operates on a ranked dissimilarity matrix rather than raw data in contrast to ANOVA. This procedure adjusts the ranging of dissimilarity to non-metric multi-dimensional scaling (NMDS) where it is possible to reduce dimension and visualize non-parametric multi-dimensional data [114,115]. They are dedicated to more complex datasets which are prone to type I error. Moreover, they allow for the calculation of differences at high-resolution taxonomic levels [31]. Principal coordinate analysis (PCoA) is a method of multidimensional visualization of similarities or dissimilarities in data. In PCoA the matrices of similarities and dissimilarities are created, subsequently each element is assigned to a location in a low-dimensional space, e.g., 3D [116]. Canonical analyzes of principle components (CAP) is a useful procedure with limited ordinances. It allows for the creation of a sensible limited ordinance based on any dissimilarity matrix or distance measure. This measure is used in multidimensional ecological data and allows for the use of any measure of distance or dissimilarity or takes into account the structure of correlation between variables in the data cloud [117].
The above-mentioned statistical and bioinformatics tools are just some of the many used in molecular biology data analysis. Their application depends on the obtained data and conducted experiment and the final effect which we want to achieve. As demonstrated by Blaud et al. [72], the results obtained by the T-RFLP approach require different multivariate analyses. Clustering or NMDS analysis is not sufficient and needs to be supplemented by statistical tests such as ANOSIM or PERMANOVA. In general, the genetic profiles obtained as a result of DGGE assay were presented in the form of UPGMA (unweighted pair group mean average) dendrogram using Dice coefficient of similarity [74,118]. On the other hand, Wang et al. [75] analyzed data obtained as a result of DGGE approach using PCA plot. Profiles resulting from the T-RFLP procedure were most often analyzed using PCA [74,119,120]. De Vrieze et al. [68] obtained profiles of microorganisms as a result of NGS and T-RFLP analyzed using NMDS plots constructed from Bray–Curtis, Chao, Jaccard, Kulczynski, and Mountford distance measures. The Bray–Curtis distance-based NMDS analysis used in the study by Tan et al. [121] allowed for comparison of bacterial and fungal communities in different soil samples, where black morel was cultivated. The community profiles were shown to be divided into two groups associated with fruiting and non-fruiting forms of black morel. Estimators of α-diversity included in the study were the Shannon-Wiener index, the inverse Simpson’s index, and Pielou’s evenness index, which allowed the assessment of diversity and evenness and were selected as candidates for potential correlation tests. The Shannon-Wiener and inverse Simpson’s diversity indices showed positive correlations with estimated yield levels. Similar correlations were also shown for Pielou’s evenness index and fungus yield. In Adhicari et al. [57] study α-diversity of soil bacterial populations in a soil sample determined by the NGS approach was expressed by using Chao1, Shannon, and Simpson indices. In contrast, β-diversity was determined using PCoA (considering Bray–Curtis dissimilarities) and UPGMA based on unweighted UniFrac distances. The analyses allowed to conclude that the samples studied were characterized by similar trends in bacterial community and diversity.
5. Genetic Diversity of Soil Microorganisms—Application Areas
5.1. Fertilization Strategies, Agricultural Soil Monitoring
Li et al. [122] investigated the effect of hydrogen gas as a by-product during the process of nitrogen fixation in Medicago sativa field soil. After DNA isolation, the bacterial 16S rDNA gene was amplified using primers targeting the V3 and V4 region (338F and 806R). After purification, DNA was sequenced using Illumina platforms. The studies showed the influence of hydrogen on the composition of the rhizosphere on M. sativa. Among the 18 soil samples, Proteobacteria and Actinobacteria dominated, with Proteobacteria being the most abundant group of bacteria. Before the soil was treated with hydrogen, Actinobacteria were dominant, but their development was inhibited. There was also a seasonal fluctuation in diversity, with the highest diversity index recorded in July. In soil samples collected in July, and September, the most dominant bacterial species were Actinobacteria (at the beginning of the experiment) and Proteobacteria. Sampling time and the presence of hydrogen gas were the greatest impact on the microbial diversity on M. sativa rhizospheric soil. It can be assumed that the tendency towards Actinobacteria and Proteobacteria is related to the oxidation of hydrogen in order to maintain trace concentrations of atmospheric hydrogen.
Gryta et al. [118] conducted study on the effect of fertilization strategies on the microorganisms biodiversity, including those involved in the transformation of nitrogen compounds (nitrification). In the experiment DGEE assay was used to determine the structure of the bacterial community, while the patterns obtained by the T-TFLP method provided the abundance of ammonia-oxidizing archaea. Soil samples were obtained from an experiment on winter wheat. The soils were enriched with dairy sewage sludge and mineral fertilizers. In general, it was found that the treatment of soil with sewage sludge similarly affects the index biodiversity of microorganisms including nitrification bacteria. It means that the application of the sludge can be an alternative or supplement to mineral fertilization.
Semenov et al. [108] was indicated, that 16S amplicon sequencing revealed that long-term fertilization may have a higher effect on rhizosphere prokaryotic communities rather than plant species. Additionally, the authors proved that NPK fertilizers caused not only a change in the structure of the rhizosphere microbiome, but also drastically reduced the number of unique RSV (by 36%) and prokaryotic diversity (from 6.5 to 5.6). Contrary, long-term manure fertilization increased significantly prokaryotic abundance and diversity due to development of many underrepresented taxa.
The metagenomics study by Sun et al. [123] showed that an excessive nitrogen (N) fertilization in agricultural ecosystems strongly influences the microbial N-cycle processes in soil. They reported that long-term nitrogen application increased the number of microorganisms involved in most nitrogen transformation processes, but reduced the number of nitrogen-fixing assemblers. Additionally, it has been investigated that different responses of taxa to nitrogen fertilization within the same functional group may be important in maintaining the microbial nitrogen cycle in complex and dynamic environments.
Akinola et al. [124] characterized functional genes of the microbial community of maize rhizosphere using shotgun metagenomics by Illumina sequencing. The β-diversity indicated that there is a great significant difference between the genes extracted from rhizosphere soil (especially Ls) compared to bulk soil. Moreover, the high relative abundance of stress-reducing genes extracted in the experiment was observed. It confirms the fact that plant rhizosphere is not only a habitat for organisms beneficial to plants, but also it is a source of bio-fertilizers.
16S and 18S rRNA gene were sequenced in study provided by Sadet-Bourgeteau et al. [125] in order to evaluate the microbial structure after different organic waste products application (co-composting of green wastes with sewage sludge and a farmyard manure). It was found that the biodiversity of prokaryotes and fungi differed depending on the type of soil and the waste used. Large changes in prokaryotic genetic structures were observed after the treatment of soil with composted green waste with sewage sludge.
5.2. Soil Contamination and Remediation Monitoring
Heavy metals are one of the most important soil contaminants. Their presence in the environment may exert negative effects, such as disruption of physiological and enzymatic functions, damage to cell membranes, disruption of their permeability or DNA degradation [126]. Heavy metals may also change soil biodiversity, as investigated by Xu et al. [127]. DNA was isolated from the soil and the 16S rRNA gene was amplified using conserved bacterium specific primers (515F and 806R). The obtained results indicated that the richness and diversity of bacteria varied significantly between samples from different geographic regions. It was showed that Zn and Pb contamination had a significant impact on the bacterial community composition in the soil. Moreover, it was reported that bacteria from the genus Verrucomicrobia and genus Bradyrhizobium were resistant to Zn and Pb and they may be useful in the bioremediation of heavy metal contaminated sites.
Zhao et al. [128] studied the effect of heavy metals (Cd, Cu, Ni, Pb, and Zn) in soils surrounding mines on the number and biodiversity of bacteria. The extracted DNA was amplified by PCR using barcode-specific primers encoding the 16S rRNA gene (515 F and 806 R). The microbial community was determined by DNA sequencing on the Illumina HiSeq platform. The sequencing 16S rRNA was used to analyze the number and structural diversity of communities’ microorganisms in soil at risk of contamination with heavy metals. It was noted that soil physicochemical properties, sampling depth and heavy metals significantly influenced the microbial community in the soil. The most dominant were Proteobacteria (41.71%) and Firmicutes (20.44%), and the correlation analysis showed that the two bacteria were positively associated with Cu, Zn, Pb. This means that Proteobacteria and Firmicutes were highly resistant to heavy metals.
Jiang et al. [129] studied the microbiome and resistors of copper open-pit mine tailing through metagenomic sequencing and taxonomic analysis. Research revealed that Actinobacteria, Proteobacteria, Acidobacteria, Euryarchaeota, and Nitrospirae were the most abundant type in this tailing. Among all detected heavy metal and antibiotic resistance genes, merA and rpoB2 were the most numerous. Additionally, the possible influence of heavy metals on the formation of the microbiome and resistance in mine tailings was indicated.
Ma et al. [130] conducted research on changes in the abundance and structure of microorganisms during bioremediation (using plant and white-root fungi) of soils contaminated with polycyclic aromatic hydrocarbons (PAHs). After extraction of soil DNA, the supersvariable V3-V4 region of the bacterial 16S rRNA gene was amplified using primers 341F/806R and then sequenced. In addition, the fungal rRNA internal transcribed spacer (ITS) region was amplified and sequenced using the ITS3_KYO2F/ITS4R primers pair. Additionally, the qPCR of 16S rDNA, ITS, and the PAH-RHDα GP gene was performed. It was found that on day 60th, remediation with plants and Crucibulum laeve achieved the highest efficiency in removing phenanthrene, pyrene and benzo(a)pyrene. Moreover, mycoremediation using C. laeve caused an increase in the relative abundance of Rhizobium and Bacillus. Additionally, it was shown that inoculation of the combination of S. viminalis and C. laeve synergistically stimulated the growth of indigenous PAH-degrading microorganisms.
Dou et al. [131] in their study used a taxonomic and functional metagenomics technique to examine the effectiveness of bioremediation of phenanthrene contaminated soil. Presented experiment was based on bacteria immobilized in layer-by-layer microcapsules. It was shown that bioaugmentation with the participation of immobilized bacteria caused significant changes in the communities of soil microorganisms contaminated with phenanthrene, favoring its degradation. The key microorganisms increasing the degradation effect caused by immobilized bacteria were the clusters of Bacteroides, Gemmatimonadetes, and Acidobacteria, as well as the genera, including Streptomyces, Ramlibacter, Mycobacterium, Phycicoccus, Gemmatirosa, Flavisolibacter, Micromonospora, Acid_Candidatus_Koribacter and Gemmatimonas.
5.3. Effect of Pesticides and Another Organic Xenobiotis
The use of pesticides is now an essential procedure that ensures and improves food production. However, it also causes unfavorable changes in the functioning of ecosystems and in shaping their biodiversity [2,132]. Therefore, scientists undertake research in which they use molecular methods to determine the effect of plant protection products on soil microflora. Du et al. [20] conducted studies on the effect of mesotrione (2-[4-(methylsulfonyl)-2-nithobenzoyl]-1, 3-cyclohexanedione) on biodiversity and abundance of soil microbiota using the t-RFLP method. After DNA extraction, the 16S rRNA gene was amplified using primers 27F and 1492R, of which forward primer was fluorescently labeled (6FAM). PCR product was purified and digested by Msp I enzyme at 37 °C by 8h. Digested product was analyzed by capillary genetic analyzer. Authors investigated also the influence of mesotrione application on quantity ammonia-oxidising archaea by real-time PCR analysis. They were using AOA-amoA and AOB-amoA genes. The conducted research showed that the number of bacteria, fungi and actinomycetes decreased in the soil mesotrione treated with the doses of 1.0 and 5.0 mg/kg. In the soil with 0.1 mg/kg of mesotrione, only the number of fungi decreased at the end of the experiment. Analysis of the t-RFLP profile revealed that mesotrione influences the structure of the soil microbial community in a dose-independent manner. Moreover, the abundance of the AOA-amoA and AOB-amoA genes decreased after mesotrione application at the doses of 1.0 and 5.0 mg/kg of soil.
Singh and Singla [133] used molecular methods to the identification of diuron-utilising bacteria. After DNA extraction, PCR of the 16S rRNA gene was conducted using universal primers (27F and 1492R). The purified product was sequenced by the dideoxy chain terminator method (Sanger sequencing). According to 16S rRNA gene sequence analysis, Bacillus sps were the endophytes having ability to degrading diuron. The isolated endophytes showed the ability to promote plant growth. Among the isolated endophytes, Bacillus licheniformis strain SDS12 showed the best efficiency in the degradation of diuron. The authors also indicated that the SDS12 strain can be used in water bodies for degradation diuron and reduction in diuron toxicity towards algae.
In study conducted by Aguiar et al. [21] metagenomic sequencing was performed in order to identify of atrazine-degrading microorganisms in rhizospheric soils. The soil genomic DNA was isolated and purified, and then the sequencing was done on the Illumina HiSeq platform. Studies have shown that Mycobacterium, Conexibacter, Bradyrhizobium, Solirubrobacter, Rhodoplanes, Streptomyces, Geothrix, Gaiella, Nitrospira and Haliangium dominated in the rhizospheric soils of Inga striata and Caesalphinea ferrea contaminated with atrazine. Moreover, in the rhizosphere of I. striata the genes atzD, atzE and atzF were detected, while in C. ferrea atzE and atzF. The work represents the first report for the species Agrobacterium rhizogenes and Candidatus Muproteobacteria bacterium and Micromonospora genera as atrazine degraders.
Serbent et al. [134] assessed the microbiota composition in an experimental rice planting and long-term pesticide application using sequencing of 16S and 18S rRNA amplicons. They examined four components of a complete agricultural system: affluent water, rice rhizosphere soil, sediment from a storage pond, and effluent water. Using the β-diversity analysis of bacterial communities, two well-defined aggregates were shown separately for water and sediment/rhizospheric soil samples. The rhizosphere and sediment were richer as compared to the effluent. In general, the rhizosphere was characterized by the greatest evenness. Contrary to bacterial communities, the diversity of microeukaryotes according to Shannon was significantly different between affluent and effluent. During mapping of the metabolic pathways, genes related to aromatic degradation were identified, including those related to pesticide degradation. The authors also showed that the effluent is a selective environment for fungi. In addition, it was investigated that overall fungal diversity was higher in affluent, i.e., in the water that reached the system before pesticide application, and that prokaryotic diversity was the lowest.
6. Conclusions
The genetic diversity of microorganisms analysis is a useful and often indispensable tool allowing to assess the impact of various pollutants such heavy metals, pesticides or PAHs on the biological condition of soils. It is also a good way to assess the performed reclamation, bioremediation and phytoremediation of degraded and chemically contaminated soil. Soil as a complex matrix requires a special approach when choosing the appropriate molecular method due to the heterogeneous and variable composition and structure, the presence of inhibitors and other compounds affecting the analysis result, as well as susceptibility to changing biotic and abiotic factors. The quality of the isolated DNA and the specificity of PCR reaction are also factors of great important.
The use of molecular techniques in an environmental biotechnology facilitates the analysis of the community of microorganisms too difficult to study with conventional laboratory methods, associated with complex biochemical profiles, problems with the use of selective and differentiating media for their incubation. Currently, the most popular methods for analyzing the soil microbiome are NGS and TGS techniques. Nevertheless, procedures based on molecular biology are a perspective and still evolving area of knowledge that is a challenge for scientists. It should be noted that molecular techniques are not limited to the work of biologists and microbiologists and require an interdisciplinary approach involving biochemists, biotechnologists and environmental engineers. As it turns out, bioinformatics and the development of statistical methods are also of great importance in the analysis of biodiversity.
Summarizing, all the above-mentioned aspects support the fact that there is a constant need to modify, modernize and develop methods and tools useful in the analysis of genetic microbiological diversity of soil. The challenge for researchers is still to improve the methods of DNA extraction and gene amplification, which constitute the basis for further analyzes of soil biodiversity and determine its quality and further success. Another important aspect, which is constantly updated and improved, is the assembly of genomes in HTS technologies, the development of bioinformatics and statistical techniques, which will inform about the biological state of the soil with even greater precision. It is also important to conduct comprehensive soil monitoring in conjunction with metagenomics, metatranscriptomics, metabolomics and metaproteomics methods, which will give a complete picture of the microbial structure-function relationship in soil. Finally, modern HTS techniques should be made more user-friendly, more common and accessible to interested researchers.
Funding
This work was financially supported by the Ministry of Education and Science, Poland, under the research project number WZ/WB-IIŚ/6/2022.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cornea, C.P.; Voaideş, C.; Ciuca, M.; Stan, V.; Gament, E.; Razec, I.; Duşa, M. Molecular Methods for Assessement the Bacterial Communities from Different Type of Soils in Romania. Not. Bot. Horti Agrobot. Cluj-Napoca 2011, 39, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Wołejko, E.; Jabłońska-Trypuć, A.; Wydro, U.; Butarewic, Z.A.; Łozowicka, B. Soil biological activity as an indicator of soil pollution with pesticides—A review. Appl. Soil. Ecol. 2020, 147, 103356. [Google Scholar] [CrossRef]
- Singh, B.K.; Campbell, C.D.; Sorenson, S.J.; Zhou, J. Soil genomics. Nat. Rev. Microbiol. 2009, 7, 756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaden, R.; Krolla-Sidenstein, P. How to Show the Real Microbial Biodiversity? A Comparison of Seven DNA Extraction Methods for Bacterial Population Analyses in Matrices Containing Highly Charged Natural Nanoparticles. Microorganisms 2015, 3, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Smalla, K.; Oros-Sichler, M.; Milling, A.; Heuer, H.; Baumgarte, S.; Becker, R.; Neuber, G.; Kropf, S.; Ulrich, A.; Tebbe, C.C. Bacterial diversity of soils assessed by DGGE, T-RFLP and SSCP fingerprints of PCR-amplified 16S rRNA gene fragments: Do the different methods provide similar results? J. Microbiol. Methods 2007, 69, 470–479. [Google Scholar] [CrossRef]
- Tanveer, A.; Yadav, S.; Yadav, D. Comparative assessment of methods for metagenomic DNA isolation from soils of different crop growing fields. 3 Biotech 2016, 6, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.K.; Singh, H.; Sharma, P.C. An improved method suitable for isolation of high-quality metagenomic DNA from diverse soils. 3 Biotech 2017, 7, 171. [Google Scholar] [CrossRef]
- Salonen, A.; Nikkilä, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilić-Stojanović, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative Analysis of Fecal DNA Extraction Methods with Phylogenetic Microarray: Effective Recovery of Bacterial and Archaeal DNA Using Mechanical Cell Lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
- Delmont, T.O.; Robe, P.; Clark, I.; Simonet, P.; Vogel, T.M. Metagenomic Comparison of Direct and Indirect Soil DNA Extraction Approaches. J. Microbiol. Methods 2011, 86, 397–400. [Google Scholar] [CrossRef]
- Mazziotti, M.; Henry, S.; Laval-Gilly, P.; Bonnefoy, A.; Falla, J. Comparison of Two Bacterial DNA Extraction Methods from Non-Polluted and Polluted Soils. Folia Microbiol. 2018, 63, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, G.; Sani, R.K. Molecular Techniques to Assess Microbial Community Structure, Function, and Dynamics in the Environment. In Microbes and Microbial Technology; Ahmad, I., Ahmad, F., Pichtel, J., Eds.; Springer: New York, NY, USA, 2011; pp. 29–57. [Google Scholar] [CrossRef]
- Abed, R.M.; Safi, N.M.D.; Köster, J.; de Beer, D.; El-Nahhal, Y.; Rullkötter, Y.; Garcia-Pichel, F. Microbial Diversity of a Heavily Polluted Microbial Mat and Its Community Changes following Degradation of Petroleum Compounds. Appl. Environ. Microbiol. 2002, 68, 1674–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, P.B.L.; Lallias, D.; Creer, S.; Seaton, F.M.; Kenny, J.G.; Eccles, R.M.; Griffiths, R.I.; Lebron, I.; Emmett, B.A.; Robinson, D.A.; et al. Divergent national-scale trends of microbial and animal biodiversity revealed across diverse temperate soil ecosystems. Nat. Commun. 2019, 10, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidstedt, M.; Rådström, P.; Hedman, J. PCR inhibition in qPCR, dPCR and MPS—mechanisms and solutions. Anal. Bioanal. Chem. 2020, 412, 2009–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenov, M.V. Metabarcoding and Metagenomics in Soil Ecology Research: Achievements, Challenges, and Prospects. Biol. Bull. Rev. 2021, 11, 40–53. [Google Scholar] [CrossRef]
- Liu, W.; Liu, L.; Yang, X.; Deng, M.; Wang, Z.; Wang, P.; Yang, S.; Li, P.; Peng, Z.; Yang, L.; et al. Long-Term Nitrogen Input Alters Plant and Soil Bacterial, but Not Fungal Beta Diversity in a Semiarid Grassland. Glob. Chang. Biol. 2021, 27, 3939–3950. [Google Scholar] [CrossRef]
- Socolar, J.B.; Gilroy, J.J.; Kunin, W.E.; Edwards, D.P. How Should Beta-Diversity Inform Biodiversity Conservation? Trends Ecol. Evol. 2016, 31, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular Ecological Network Analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Zhang, W.; Zhong, Z.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Differential responses of soil microbial biomass, diversity, and compositions to altitudinal gradients depend on plant and soil characteristics. Sci. Total Environ. 2018, 610–611, 750–758. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Souza, M.D.F.; de Laia, M.L.; Melo, J.D.O.; da Costa, M.R.; Gonçalves, J.F.; Silva, D.V.; dos Santos, J.B. Metagenomic analysis reveals mechanisms of atrazine biodegradation promoted by tree species. Environ. Pollut. 2020, 267, 115636. [Google Scholar] [CrossRef]
- García-Lozano, M.; Hernández-De Lira, I.O.; Huber, D.H.; Balagurusamy, N. Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes 2019, 7, 408. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhu, Y.; Zhu, L.; Zhang, J.; Li, B.; Wang, J.; Wang, J.; Zhang, C.; Cheng, C. Effects of the herbicide mesotrione on soil enzyme activity and microbial communities. Ecotoxicol. Environ. Saf. 2018, 164, 571–578. [Google Scholar] [CrossRef]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 2020, 61, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Genet. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Bhadury, P. Methods of Assessment of Microbial Diversity in Natural Environments. In Microbial Diversity in the Genomic Era, 1st ed.; Das, S., Dash, H., Eds.; Academic Press, Elsevier: London, UK, 2019; pp. 3–14. [Google Scholar] [CrossRef]
- Kumar, R.; Joshi, S.R. Microbial Ecology of Soil: Studying the diversity of microorgansims in the most complex of the environments. A review. Adv. Appl. Microbiol. 2015, 19, 267–279. [Google Scholar]
- Nannipieri, P.; Penton, C.R.; Purahong, W.; Schloter, M.; van Elsas, J.D. Recommendations for soil microbiome analyses. Biol. Fertil. Soils 2019, 55, 765–766. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.K.; Tripathi, V.; Prabha, R.; Chaurasia, R.; Singh, D.P.; Rao, C.S.; El-Keblawy, A.; Abhilash, P.C. Bioinformatics Tools for Soil Microbiome Analysis. In Unravelling the Soil Microbiome, 1st ed.; SpringerBriefs in Environmental Science; Springer: Cham, Switzerland, 2020; pp. 61–70. [Google Scholar] [CrossRef]
- Niu, S.-Y.; Yang, J.; McDermaid, A.; Zhao, J.; Kang, Y.; Ma, Q. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Briefings Bioinform. 2017, 19, 1415–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, C.; Sadowsky, M.J. Practical considerations for sampling and data analy-sis in contemporary metagenomics-based environmental studies. J. Microbiol. Methods 2018, 154, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA Recovery from Soils of Diverse Composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, Y. Improvements in Extraction Methods of High-molecular-weight DNA from Soils by Modifying Cell Lysis Conditions and Reducing Adsorption of DNA onto Soil Particles. Microbes Environ. 2021, 36, ME21017. [Google Scholar] [CrossRef]
- Philippot, L.; Abbate, C.; Bispo, A.; Chesnot, T.; Hallin, S.; Lemanceau, P.; Lindström, K.; Pandard, P.; Romero, E.; Schloter, M.; et al. Soil microbial diversity: An ISO standard for soil DNA extraction. J. Soils Sediments 2010, 10, 1344–1345. [Google Scholar] [CrossRef]
- Young, J.M.; Rawlence, N.J.; Weyrich, L.S.; Cooper, A. Limitations and Recommendations for Successful DNA Extraction from Forensic Soil Samples: A Review. Sci. Justice 2014, 54, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Fatima, F.; Pathak, N.; Rastogi Verma, S. An Improved Method for Soil DNA Extraction to Study the Microbial Assortment within Rhizospheric Region. Mol. Biol. Int. 2014, 2014, 518960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauth, S.; Schmidt, H.; Tippkötter, R. Comparison of Commercial Kits for the Extraction of DNA from Paddy Soils. Lett. Appl. Microbiol. 2013, 56, 222–228. [Google Scholar] [CrossRef]
- Felczykowska, A.; Krajewska, A.; Zielińska, S.; Łoś, J.M. Sampling, Metadata and DNA Extraction—Important Steps in Metagenomic Studies. Acta Biochim. Pol. 2015, 62, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Kuske, C.R.; Banton, K.L.; Adorada, D.L.; Stark, P.C.; Hill, K.K.; Jackson, P.J. Small-Scale DNA Sample Preparation Method for Field PCR Detection of Microbial Cells and Spores in Soil. Appl. Environ. Microbiol. 1998, 64, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Krsek, M.; Wellington, E.M. Comparison of Different Methods for the Isolation and Purification of Total Community DNA from Soil. J. Microbiol. Methods 1999, 39, 1–16. [Google Scholar] [CrossRef]
- Islam, M.R.; Sultana, T.; Melvin Joe, M.; Cho, J.-C.; Sa, T. Comparisons of Direct Extraction Methods of Microbial DNA from Different Paddy Soils. Saudi J. Biol. Sci. 2012, 19, 337–342. [Google Scholar] [CrossRef]
- Gaur, M.; Vasudeva, A.; Singh, A.; Sharma, V.; Khurana, H.; Negi, R.K.; Lee, J.-K.; Kalia, V.C.; Misra, R.; Singh, Y. Comparison of DNA Extraction Methods for Optimal Recovery of Metagenomic DNA from Human and Environmental Samples. Indian J. Microbiol. 2019, 59, 482–489. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Olson, B.H. Rapid Method for Direct Extraction of DNA from Soil and Sediments. Appl. Environ. Microbiol. 1991, 57, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Volossiouk, T.; Robb, E.J.; Nazar, R.N. Direct DNA Extraction for PCR-Mediated Assays of Soil Organisms. Appl. Environ. Microbiol. 1995, 61, 3972–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.; Satyanarayana, T. An Improved Protocol for DNA Extraction from Alkaline Soil and Sediment Samples for Constructing Metagenomic Libraries. Appl. Biochem. Biotechnol. 2011, 165, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Devi, T.; Verma, V.; Rasool, S. Comparative Studies on the Extraction of Metagenomic DNA from Various Soil and Sediment Samples of Jammu and Kashmir Region in Prospect for Novel Biocatalysts. IOSR J. Environ. Sci. Toxicol. Food Technol. 2014, 8, 46–56. [Google Scholar] [CrossRef]
- Martin-Laurent, F.; Philippot, L.; Hallet, S.; Chaussod, R.; Germon, J.C.; Soulas, G.; Catroux, G. DNA Extraction from Soils: Old Bias for New Microbial Diversity Analysis Methods. Appl. Environ. Microbiol. 2001, 67, 4397. [Google Scholar] [CrossRef] [Green Version]
- Petric, I.; Philippot, L.; Abbate, C.; Bispo, A.; Chesnot, T.; Hallin, S.; Laval, K.; Lebeau, T.; Lemanceau, P.; Leyval, C.; et al. Inter-Laboratory Evaluation of the ISO Standard 11063 “Soil Quality—Method to Directly Extract DNA from Soil Samples”. J. Microbiol. Methods 2011, 84, 454–460. [Google Scholar] [CrossRef]
- Técher, D.; Martinez-Chois, C.; D’Innocenzo, M.; Laval-Gilly, P.; Bennasroune, A.; Foucaud, L.; Falla, J. Novel Perspectives to Purify Genomic DNA from High Humic Acid Content and Contaminated Soils. Sep. Purif. Technol. 2010, 75, 81–86. [Google Scholar] [CrossRef]
- Santos, S.S.; Nielsen, T.K.; Hansen, L.H.; Winding, A. Comparison of Three DNA Extraction Methods for Recovery of Soil Protist DNA. J. Microbiol. Methods 2015, 115, 13–19. [Google Scholar] [CrossRef]
- Plassart, P.; Terrat, S.; Thomson, B.; Griffiths, R.; Dequiedt, S.; Lelievre, M.; Regnier, T.; Nowak, V.; Bailey, M.; Lemanceau, P.; et al. Evaluation of the ISO Standard 11063 DNA Extraction Procedure for Assessing Soil Microbial Abundance and Community Structure. PLoS ONE 2012, 7, e44279. [Google Scholar] [CrossRef]
- Basim, Y.; Mohebali, G.; Jorfi, S.; Nabizadeh, R.; Ghadiri, A.; Moghadam, M.A.; Soleymani, F.; Fard, N.J.H. Comparison of Performance and Efficiency of Four Methods to Extract Genomic DNA from Oil Contaminated Soils in Southwestern of Iran. J. Environ. Health Sci. Eng. 2020, 18, 463–468. [Google Scholar] [CrossRef]
- Bollmann-Giolai, A.; Giolai, M.; Heavens, D.; Macaulay, I.; Malone, J.; Clark, M.D. A Low-Cost Pipeline for Soil Microbiome Profiling. Microbiologyopen 2020, 9, e1133. [Google Scholar] [CrossRef]
- Leite, D.C.A.; Balieiro, F.C.; Pires, C.A.; Madari, B.E.; Rosado, A.S.; Coutinho, H.L.C.; Peixoto, R.S. Comparison of DNA Extraction Protocols for Microbial Communities from Soil Treated with Biochar. Braz. J. Microbiol. 2014, 45, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürgmann, H.; Pesaro, M.; Widmer, F.; Zeyer, J. A Strategy for Optimizing Quality and Quantity of DNA Extracted from Soil. J. Microbiol. Methods 2001, 45, 7–20. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, K.D.; Yilmaz-Swenson, T.; Elsayed, N.M.; Cruz, D.A.; Rodriguez, R.R.; Kritzer, M.D.; Peterchev, A.V.; Gray, M.; Lewis, S.R.; Roach, J.; et al. Comparative Evaluation of a New Magnetic Bead-Based DNA Extraction Method from Fecal Samples for Downstream next-Generation 16S RRNA Gene Sequencing. PLoS ONE 2018, 13, e0202858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, M.; Kim, S.W.; Kim, H.S.; Kim, K.Y.; Park, H.B.; Kim, K.J.; Lee, Y.S. Bacterial Community and Diversity from the Watermelon Cultivated Soils through next Generation Sequencing Approach. Plant Pathol. J. 2021, 37, 521–532. [Google Scholar] [CrossRef]
- Wüst, P.K.; Nacke, H.; Kaiser, K.; Marhan, S.; Sikorski, J.; Kandeler, E.; Daniel, R.; Overmann, J. Estimates of Soil Bacterial Ribosome Content and Diversity Are Significantly Affected by the Nucleic Acid Extraction Method Employed. Appl. Environ. Microbiol. 2016, 82, 2595–2607. [Google Scholar] [CrossRef] [Green Version]
- Sagova-Mareckova, M.; Cermak, L.; Novotna, J.; Plhackova, K.; Forstova, J.; Kopecky, J. Innovative Methods for Soil DNA Purification Tested in Soils with Widely Differing Characteristics. Appl. Environ. Microbiol. 2008, 74, 2902–2907. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.S.; Nunes, I.; Nielsen, T.K.; Jacquiod, S.; Hansen, L.H.; Winding, A. Soil DNA Extraction Procedure Influences Protist 18S RRNA Gene Community Profiling Outcome. Protist 2017, 168, 283–293. [Google Scholar] [CrossRef]
- Morita, H.; Akao, S. The Effect of Soil Sample Size, for Practical DNA Extraction, on Soil Microbial Diversity in Different Taxonomic Ranks. PLoS ONE 2021, 16, e0260121. [Google Scholar] [CrossRef]
- Rousk, J.; Brookes, P.C.; Bååth, E. Contrasting Soil PH Effects on Fungal and Bacterial Growth Suggest Functional Redundancy in Carbon Mineralization. Appl. Environ. Microbiol. 2009, 75, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil Bacterial and Fungal Communities across a PH Gradient in an Arable Soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Guerrieri, A.; Bonin, A.; Münkemüller, T.; Gielly, L.; Thuiller, W.; Francesco Ficetola, G. Effects of Soil Preservation for Biodiversity Monitoring Using Environmental DNA. Mol. Ecol. 2021, 30, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Stach, J.E.M.; Bathe, S.; Clapp, J.P.; Burns, R.G. PCR-SSCP Comparison of 16S RDNA Sequence Diversity in Soil DNA Obtained Using Different Isolation and Purification Methods. FEMS Microbiol. Ecol. 2001, 36, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, L.M.; Sul, W.J.; Blackwood, C.B. Assessment of Bias Associated with Incomplete Extraction of Microbial DNA from Soil. Appl. Environ. Microbiol. 2009, 75, 5428–5433. [Google Scholar] [CrossRef] [Green Version]
- Zielińska, S.; Radkowski, P.; Blendowska, A.; Ludwig-Gałęzowska, A.; Łoś, J.M.; Łoś, M. The Choice of the DNA Extraction Method May Influence the Outcome of the Soil Microbial Community Structure Analysis. MicrobiologyOpen 2017, 6, e00453. [Google Scholar] [CrossRef] [Green Version]
- De Vrieze, J.; Ijaz, U.Z.; Saunders, A.M.; Theuerl, S. Terminal Restriction Fragment Length Polymorphism Is an “Old School” Reliable Technique for Swift Microbial Community Screening in Anaerobic Digestion. Sci. Rep. 2018, 8, 16818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valášková, V.; Baldrian, P. Denaturing Gradient Gel Electrophoresis as a Fingerprinting Method for the Analysis of Soil Microbial Communities. Plant Soil Environ. 2009, 55, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, C.; García-Cantalejo, J.; Botías, P.; Costa, G.; Nande, M.; Martin, M. New Insights into the Impact of NZVI on Soil Microbial Biodiversity and Functionality. J. Environ. Sci. Health Part A 2019, 54, 157–167. [Google Scholar] [CrossRef]
- Liu, W.T.; Marsh, T.L.; Cheng, H.; Forney, L.J. Characterization of Microbial Diversity by Determining Terminal Restriction Fragment Length Polymorphisms of Genes Encoding 16S RRNA. Appl. Environ. Microbiol. 1997, 63, 4516–4522. [Google Scholar] [CrossRef]
- Blaud, A.; Diouf, F.; Herrmann, A.M.; Lerch, T.Z. Analysing the Effect of Soil Organic Matter on Bacterial Communities Using T-RFLP Fingerprinting: Different Methods, Different Stories? Biol. Fertil. Soils 2015, 51, 959–971. [Google Scholar] [CrossRef] [Green Version]
- van Dorst, J.; Bissett, A.; Palmer, A.S.; Brown, M.; Snape, I.; Stark, J.S.; Raymond, B.; McKinlay, J.; Ji, M.; Winsley, T.; et al. Community Fingerprinting in a Sequencing World. FEMS Microbiol. Ecol. 2014, 89, 316–330. [Google Scholar] [CrossRef]
- Gao, G.; Yin, D.; Chen, S.; Xia, F.; Yang, J.; Li, Q.; Wang, W. Effect of Biocontrol Agent Pseudomonas Fluorescens 2P24 on Soil Fungal Community in Cucumber Rhizosphere Using T-RFLP and DGGE. PLoS ONE 2012, 7, e31806. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, T.; Zhu, L.; Wang, X.; Wang, J.; Wang, J.; Du, Z.; Li, B. Effects of Successive Metalaxyl Application on Soil Microorganisms and the Residue Dynamics. Ecol. Indic. 2019, 103, 194–201. [Google Scholar] [CrossRef]
- Gryta, A.; Frąc, M. Methodological Aspects of Multiplex Terminal Restriction Fragment Length Polymorphism-Technique to Describe the Genetic Diversity of Soil Bacteria, Archaea and Fungi. Sensors 2020, 20, 3292. [Google Scholar] [CrossRef] [PubMed]
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities along an Altitudinal Gradient in Alpine Forest Soils: What Are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Osborn, A.M. Advantages and Limitations of Quantitative PCR (Q-PCR)-Based Approaches in Microbial Ecology: Application of Q-PCR in Microbial Ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Comparison of Droplet Digital PCR and Quantitative Real-Time PCR for Examining Population Dynamics of Bacteria in Soil. Appl. Microbiol. Biotechnol. 2014, 98, 6105–6113. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hid-dessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Muhae-Ud-Din, G.; Liu, T.; Chen, W.; Zhang, J.; Gao, L. Development of the Droplet Digital PCR to Detect the Teliospores of Tilletia Controversa Kühn in the Soil with Greatly Enhanced Sensitivity. Front. Microbiol. 2020, 11, 4. [Google Scholar] [CrossRef]
- Voegel, T.M.; Larrabee, M.M.; Nelson, L.M. Development of Droplet Digital PCR Assays to Quantify Genes Involved in Nitrification and Denitrification, Comparison with Quantitative Real-Time PCR and Validation of Assays in Vineyard Soil. Can. J. Microbiol. 2021, 67, 174–187. [Google Scholar] [CrossRef]
- Prosser, J.I. Dispersing Misconceptions and Identifying Opportunities for the Use of “omics” in Soil Microbial Ecology. Nat. Rev. Microbiol. 2015, 13, 439–446. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáenz, J.S.; Roldan, F.; Junca, H.; Arbeli, Z. Effect of the Extraction and Purification of Soil DNA and Pooling of PCR Amplification Products on the Description of Bacterial and Archaeal Communities. J. Appl. Microbiol. 2019, 126, 1454–1467. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yu, Y.; Li, H.; He, J.; Lee, S.H.; Sun, K. Phylogenetic Diversity of Planktonic Bacteria in the Chukchi Borderland Region in Summer. Hai Yang Xue Bao 2013, 32, 66–74. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The Bioinformatics Tools for the Genome Assembly and Analysis Based on Third-Generation Sequencing. Brief. Funct. Genom. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Patrick, K. 454 Life Sciences: Illuminating the Future of Genome Sequencing and Personalized Medicine. Yale J. Biol. Med. 2007, 80, 191–194. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illumina Sequencing Technology Highest Data Accuracy, Simple Workflow, and a Broad Range of Applications. Available online: https://www.illumina.com/documents/products/techspotlights/techspotlight_sequencing.pdf (accessed on 23 October 2022).
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance Comparison of Illumina and Ion Torrent Next-Generation Sequencing Platforms for 16S RRNA-Based Bacterial Community Profiling. Appl. Environ. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The Impact of Next-Generation Sequencing on Genomics. J. Genet. Genom. 2011, 38, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Can, T. Introduction to Bioinformatics. In miRNomics: MicroRNA Biology and Computational Analysis; Humana Press: Totowa, NJ, USA, 2014; pp. 51–71. [Google Scholar]
- Lesk, A. Introduction to Bioinformatics, 5th ed.; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Ejigu, G.F.; Jung, J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology 2020, 9, 295. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Hupfauf, S.; Etemadi, M.; Fernández-Delgado Juárez, M.; Gómez-Brandón, M.; Insam, H.; Podmirseg, S.M. CoMA—An Intuitive and User-Friendly Pipeline for Amplicon-Sequencing Data Analysis. PLoS ONE 2020, 15, e0243241. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing Bioinformatic Pipelines for Microbial 16S RRNA Amplicon Sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef]
- Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining Operational Taxonomic Units Using DNA Barcode Data. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 1935–1943. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-Throughput Amplicon Sequencing of the Full-Length 16S RRNA Gene with Single-Nucleotide Resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenov, M.V.; Krasnov, G.S.; Semenov, V.M.; van Bruggen, A.H.C. Long-Term Fertilization Rather than Plant Species Shapes Rhizosphere and Bulk Soil Prokaryotic Communities in Agroecosystems. Appl. Soil Ecol. 2020, 154, 103641. [Google Scholar] [CrossRef]
- de la Fuente, G.; Belanche, A.; Girwood, S.E.; Pinloche, E.; Wilkinson, T.; Newbold, C.J. Pros and Cons of Ion-Torrent next Generation Sequencing versus Terminal Restriction Fragment Length Polymorphism T-RFLP for Studying the Rumen Bacterial Community. PLoS ONE 2014, 9, e101435. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M data management and analysis system v.6.0: New tools and advanced capabilities. Nucleic Acids Res. 2020, 49, D751–D763. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Beisel, J.-N.; Usseglio-Polatera, P.; Bachmann, V.; Moreteau, J.-C. A Comparative Analysis of Evenness Index Sensitivity. Int. Rev. Hydrobiol. 2003, 88, 3–15. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-Parametric Multivariate Analyses of Changes in Community Structure. Austral. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- ANOSIM: Analysis of Similarities. Available online: https://rdrr.io/rforge/vegan/man/anosim.html (accessed on 19 October 2022).
- Principal Coordinate Analysis and Non-Metric Multidimensional Scaling. In Statistics for Biology and Health; Springer: New York, NY, USA, 2007; pp. 259–264.
- Anderson, M.J.; Willis, T.J. Canonical Analysis of Principal Coordinates: A Useful Method of Constrained Ordination for Ecology. Ecology 2003, 84, 511–525. [Google Scholar] [CrossRef]
- Gryta, A.; Frąc, M.; Oszust, K. Community Shift in Structure and Functions across Soil Profile in Response to Organic Waste and Mineral Fertilization Strategies. Appl. Soil Ecol. 2019, 143, 55–60. [Google Scholar] [CrossRef]
- Ondreičková, K.; Gubišová, M.; Piliarová, M.; Horník, M.; Matušinský, P.; Gubiš, J.; Klčová, L.; Hudcovicová, M.; Kraic, J. Responses of Rhizosphere Fungal Communities to the Sewage Sludge Application into the Soil. Microorganisms 2019, 7, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čuhel, J.; Malý, S.; Královec, J. Shifts and recovery of soil microbial communities in a 40-year field trial under mineral fertilization. Pedobiologia 2019, 77, 150575. [Google Scholar] [CrossRef]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel Production Related to Soil Microbial Diversity and Evenness. Microbiol. Spectr. 2021, 9, e0022921. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Liu, R.; Li, L.; Wang, L.; Wang, W. Insight into Bacterial Community Diversity and Monthly Fluctuations of Medicago Sativa Rhizosphere Soil in Response to Hydrogen Gas Using Illumina High-Throughput Sequencing. Curr. Microbiol. 2018, 75, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, F.; Hu, C.; Liu, B. Metagenomics Reveals Taxon-Specific Responses of the Nitrogen-Cycling Microbial Community to Long-Term Nitrogen Fertilization. Soil Biol. Biochem. 2021, 156, 108214. [Google Scholar] [CrossRef]
- Akinola, S.A.; Ayangbenro, A.S.; Babalola, O.O. The Diverse Functional Genes of Maize Rhizosphere Microbiota Assessed Using Shotgun Metagenomics. J. Sci. Food Agric. 2021, 101, 3193–3201. [Google Scholar] [CrossRef]
- Sadet-Bourgeteau, S.; Houot, S.; Karimi, B.; Mathieu, O.; Mercier, V.; Montenach, D.; Morvan, T.; Sappin-Didier, V.; Watteau, F.; Nowak, V.; et al. Microbial Communities from Different Soil Types Respond Differently to Organic Waste Input. Appl. Soil Ecol. 2019, 143, 70–79. [Google Scholar] [CrossRef]
- Wydro, U.; Jabłońska-Trypuć, A.; Hawrylik, E.; Butarewicz, A.; Rodziewicz, J.; Janczukowicz, W.; Wołejko, E. Heavy Metals Behavior in Soil/Plant System after Sewage Sludge Application. Energies 2021, 14, 1584. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Z.; Hu, S.; Ruan, Z.; Jiang, J.; Chen, C.; Shen, Z. Response of Soil Bacterial Communities to Lead and Zinc Pollution Revealed by Illumina MiSeq Sequencing Investigation. Environ. Sci. Pollut. Res. Int. 2017, 24, 666–675. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, J.; Lu, J.; Sun, Y. Study on the Influence of Soil Microbial Community on the Long-Term Heavy Metal Pollution of Different Land Use Types and Depth Layers in Mine. Ecotoxicol. Environ. Saf. 2019, 170, 218–226. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, W.; Xu, H.; Cui, X.; Li, J.; Chen, J.; Zheng, B. Characterizations of Heavy Metal Contamination, Microbial Community, and Resistance Genes in a Tailing of the Largest Copper Mine in China. Environ. Pollut. 2021, 280, 116947. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, X.; Liu, J.; Cheng, Y.; Zou, J.; Zhai, F.; Sun, Z.; Han, L. Soil Microbial Community Succession and Interactions during Combined Plant/White-Rot Fungus Remediation of Polycyclic Aromatic Hydrocarbons. Sci. Total Environ. 2021, 752, 142224. [Google Scholar] [CrossRef] [PubMed]
- Dou, R.; Sun, J.; Lu, J.; Deng, F.; Yang, C.; Lu, G.; Dang, Z. Bacterial Communities and Functional Genes Stimulated during Phenanthrene Degradation in Soil by Bio-Microcapsules. Ecotoxicol. Environ. Saf. 2021, 212, 111970. [Google Scholar] [CrossRef] [PubMed]
- Łozowicka, B.; Wołejko, E.; Kaczyński, P.; Konecki, R.; Iwaniuk, P.; Drągowski, W.; Łozowicki, J.; Tujtebajeva, G.; Wydro, U.; Jablońska-Trypuć, A. Effect of Microorganism on Behaviour of Two Commonly Used Herbicides in Wheat/Soil System. Appl. Soil Ecol. 2021, 162, 103879. [Google Scholar] [CrossRef]
- Singh, A.K.; Singla, P. Biodegradation of Diuron by Endophytic Bacillus Licheniformis Strain SDS12 and Its Application in Reducing Diuron Toxicity for Green Algae. Environ. Sci. Pollut. Res. Int. 2019, 26, 26972–26981. [Google Scholar] [CrossRef]
- Serbent, M.P.; Dos Anjos Borges, L.G.; Quadros, A.; Marconatto, L.; Tavares, L.B.B.; Giongo, A. Prokaryotic and Microeukaryotic Communities in an Experimental Rice Plantation under Long-Term Use of Pesticides. Environ. Sci. Pollut. Res. Int. 2021, 28, 2328–2341. [Google Scholar] [CrossRef]
Figure 1.
The most popular commercial DNA extraction kits used in the analysis of soil genetic diversity. The sample size, material type, lysis strategy, targeting microorganism and downstream application recommended by the manufacturers were considered.
Figure 1.
The most popular commercial DNA extraction kits used in the analysis of soil genetic diversity. The sample size, material type, lysis strategy, targeting microorganism and downstream application recommended by the manufacturers were considered.
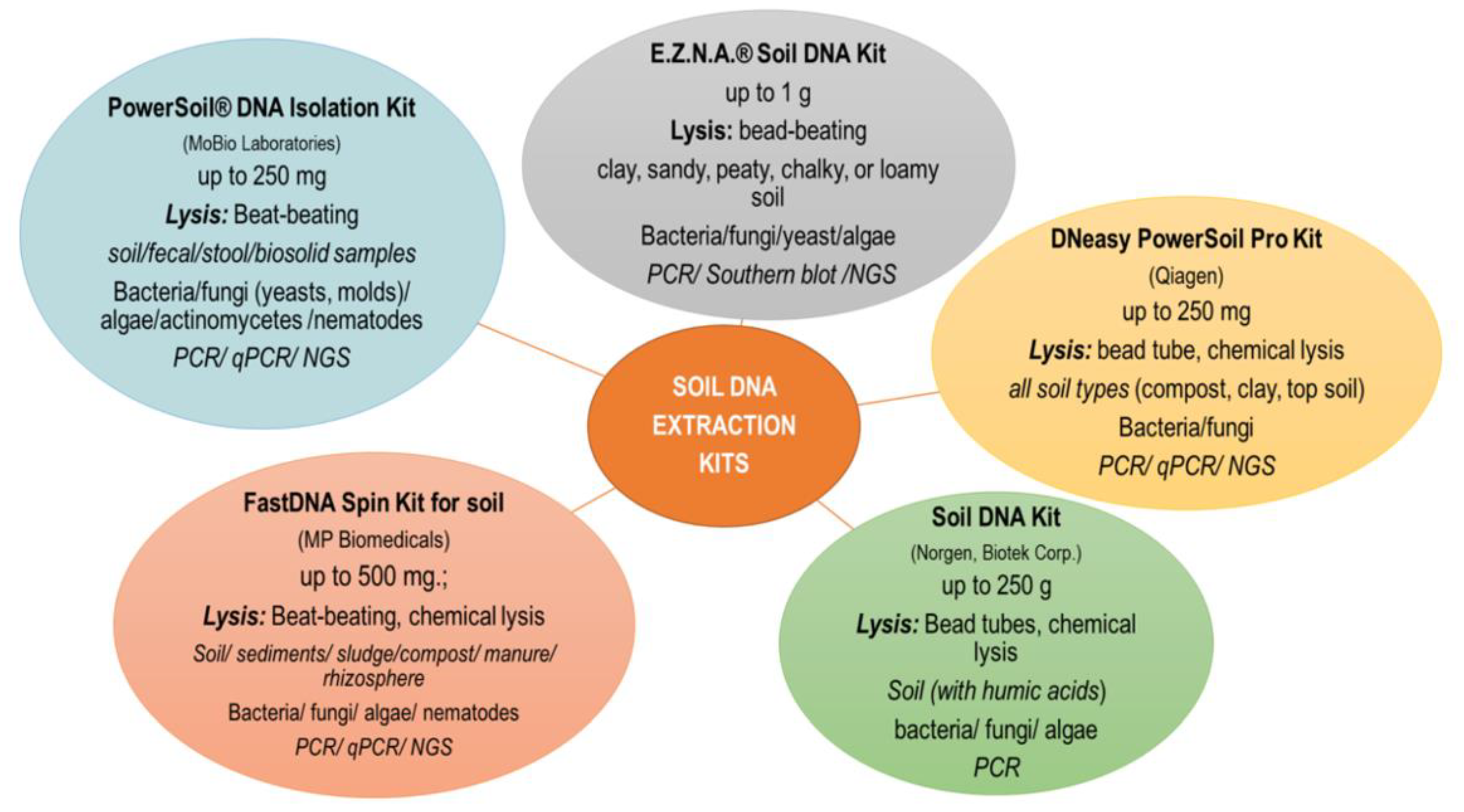
Figure 2.
The main methods of studying the diversity and structure of soil microorganisms based on DNA analysis.
Figure 2.
The main methods of studying the diversity and structure of soil microorganisms based on DNA analysis.
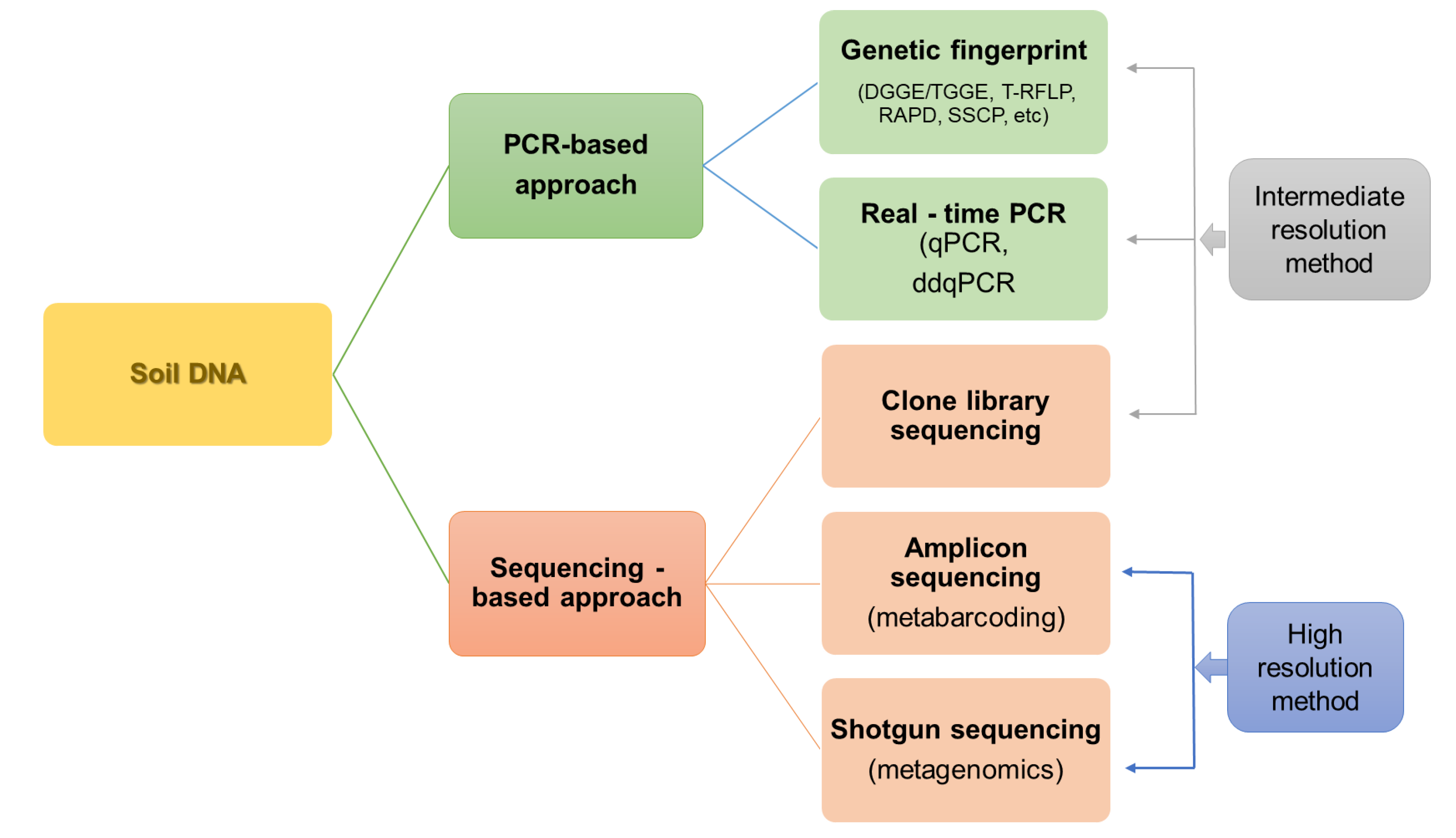
Figure 3.
Examples of metrics characteristic for α- and β-diversity.


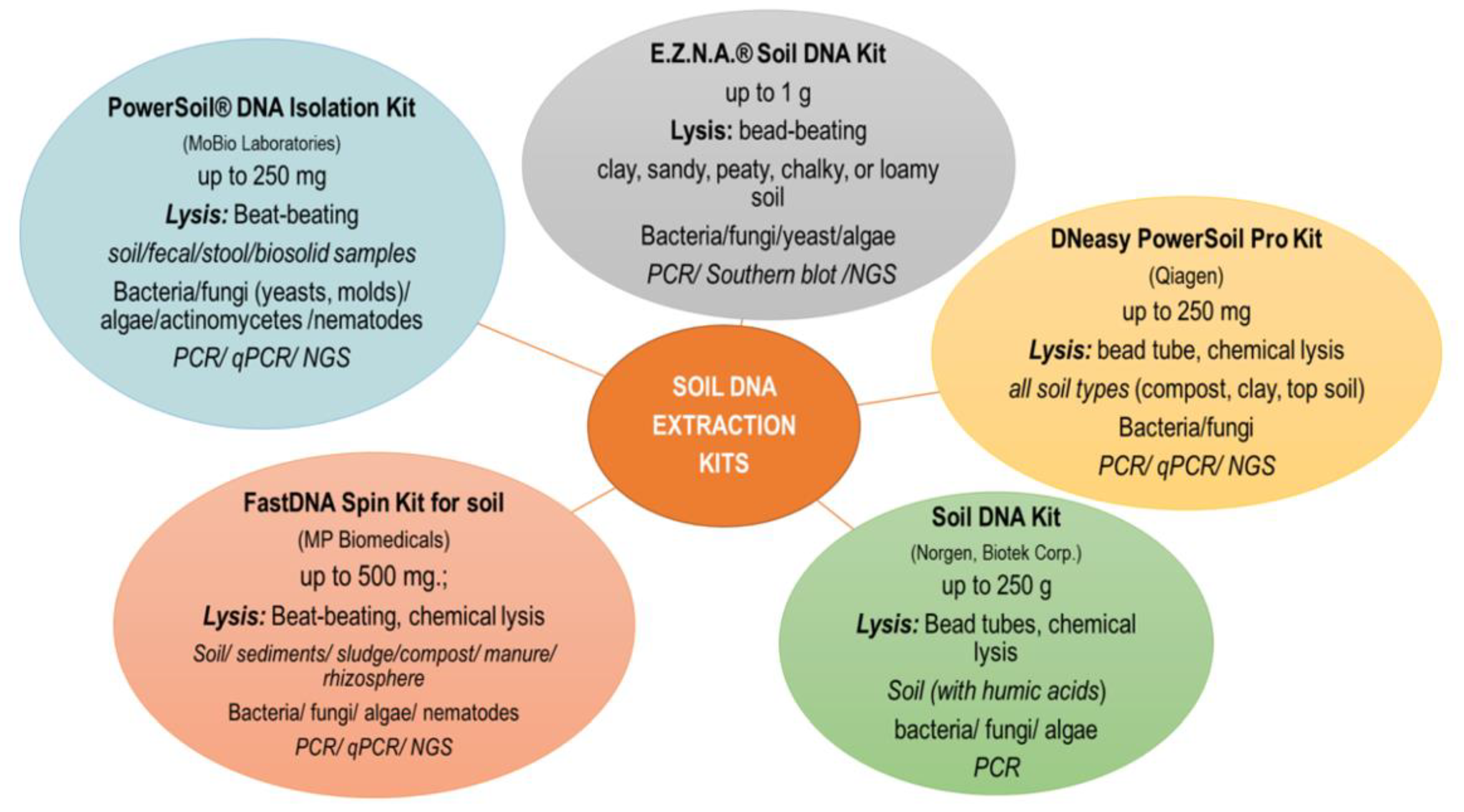{kind=link}
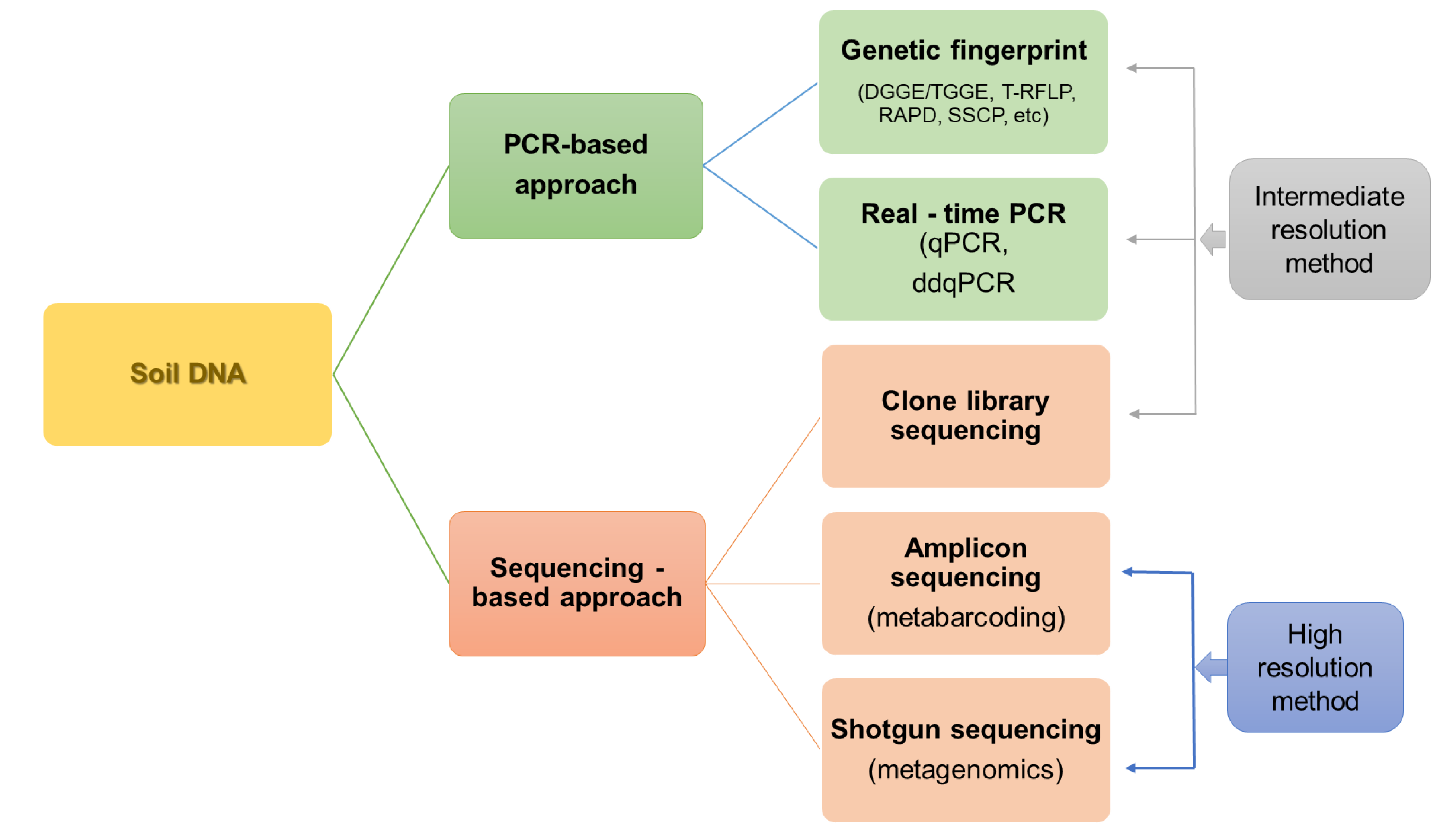{kind=link}
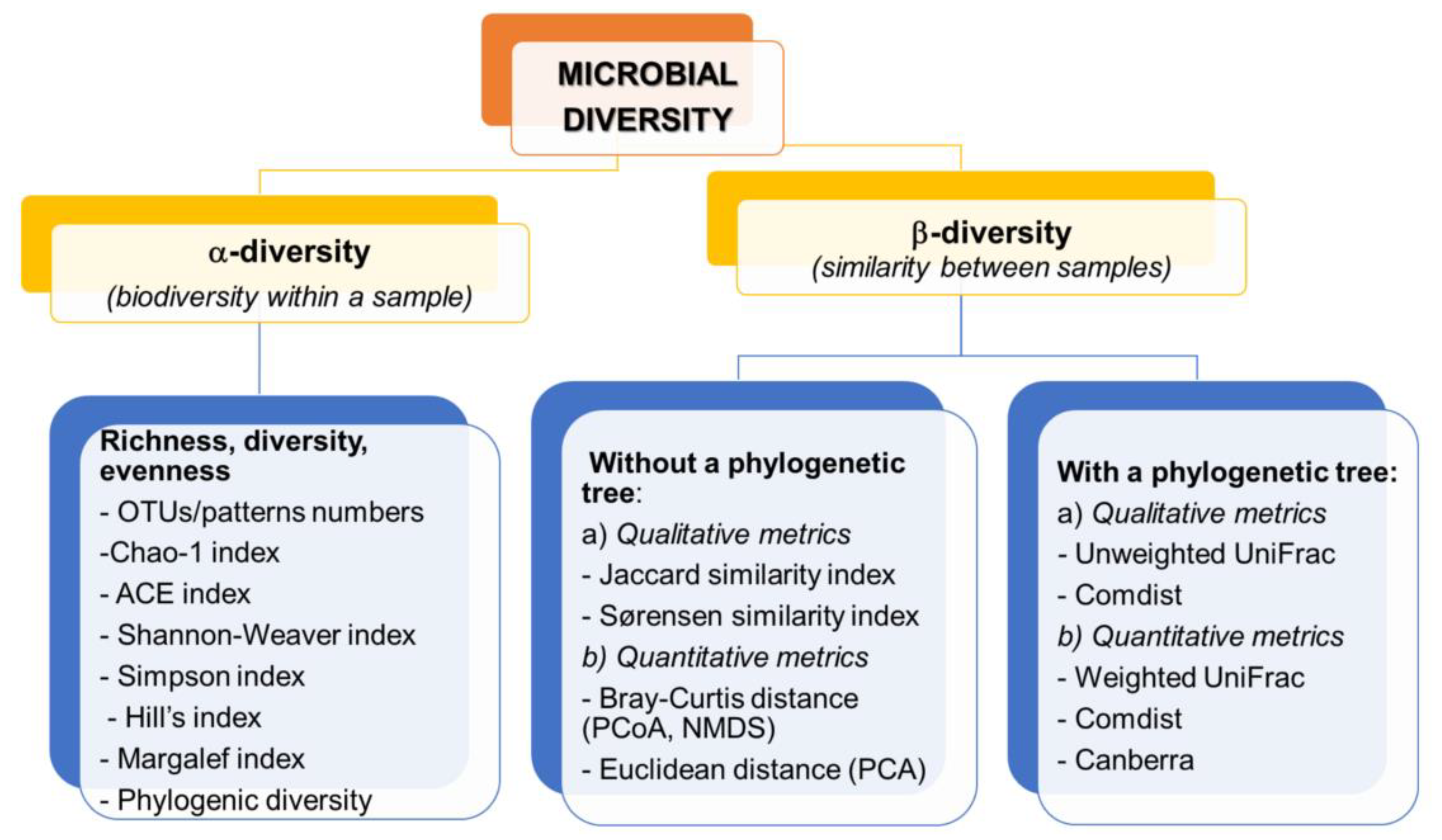{kind=link}
Table 1.
Comparison of different soil DNA extraction methods.
| References | Compared Procedures | Amount of DNA | Purity of DNA A260/280/A260/230 | Comments |
|---|---|---|---|---|
| [42] | manual lysozyme/phenol–chloroform method | 430.85 ng/µL | 1.665/1.24 | Incubation with lysozyme at 60 °C for 1 h, addition of RNase at 37 °C for 30 min and SDS |
| Qiagen DNeasy Blood & Tissue Kit | 561.3 ng/µL | 1.915/1.55 | sample was incubated at 75 °C for 10 min, silica-column based adsorption of DNA | |
| QIAamp DNA Stool mini kit | 76.35 ng/µL | 1.66/0.97 | sample was incubated at 75 °C for 10 min, silica-column based adsorption of DNA | |
| [36] | Polyethylene Glycol (PEG)/NaCl Method | 0.73 µg/mL | 1.26/1.12 | Lysis with liquid nitrogen; not suitable for PCR amplification for fungal study |
| Soil Master DNA extraction kit; | 0.79 µg/ mL | 1.32/1.21 | Lysis with liquid nitrogen; not suitable for PCR amplification for fungal study | |
| mannitol-PBS-PEG/NaCl | 2.20 µg/ mL | 1.81 /1.84 | recovery of high molecular weight soil DNA | |
| Mannitol-PBS-PCI | 2.36 µg/ mL | 1.84/1.93 | recovery of high molecular weight soil DNA | |
| Mannitol-PBS-CTAB | 2.67 µg/ mL | 1.85/2.07 | recovery of high molecular weight soil DNA | |
| [8] | Tsai and Olson method [43] | 7.55 μg/g of soil | 1.18/0.82 | physical cell lysis by repeated freeze–thaw cycles |
| Volossiouk et al. method (1995) [44] | 9.36 μg/g of soil | 1.11/0.85 | physical cell lysis by crushing soil in liquid nitrogen with freeze–thaw process; extraction cells with SDS-phenol and collected by ethanol precipitation | |
| Extraction with CTAB buffer [32] | 19.1 μg/g of soil | 1.25/0.94 | CTAB extraction buffer method; successful extraction of high-molecular weight metagenomic DNA | |
| Verma and Satyanarayana method [45] | 11.23 μg/g of soil | 1.48/1.32 | focused on cell lysis and DNA purification steps | |
| Singh et al. method [46] | 1.33 μg/g of soil | 1.02/1.00 | Extraction with Chloroform: Isoamyl alcohol (24:1); DNA precipitation with 50% PEG and 0.1 volume of 1M NaCl at −20 °C for 1h; removal of humic using CTAB extraction buffer, DNA precipitation by PEG/NaCl followed by DNA purification using 2% (w/v) CaCl2 solution; extraction of high-molecular weight, | |
| improved method | 15.55 μg/g of soil | 1.74/1.70 | using of enzymatic (lysozyme and proteinase K) and chemical (CTAB and CaCl2) strategies for cell lysis; precipitation with PEG (polyethylene glycol) and isopropanol | |
| [11] | Martin-Laurent et al.method [47] with modification by Petric et al. method [48] | 1.51–3.42 μg/g dry soil | 1.48–1.79/0.21–0.36 | Using glass beads of different sizes (300 mg of 106-μm-diameter glass beads and 2 of 2 mm diameter); 10 min incubation with potassium acetate (protein removing) |
| Técher et al. method [49] | 2.23–10.70 μg/g dry soil | 0.86–4.72/0.50–0.68 | Sand of different sizes (500 mg of fine sand and 200 mg of quartz sand); vitamin mix was used to humic acids precipitation | |
| [37] | NucleoSpin® soil kit | 0.73–2.54 μg/g of soil | 0.43–0.83/1.30–1.59 | Contain two different lysis buffers to optimize the extraction efficiency according to the soil properties, bead-beating(ceramic beads) |
| innuSPEED soil DNA kit | n.d | n.d | Lysis: Lysis tube B (2 mL) heat treatment (95–98 °C, 20 min), mechanical lysis via glass beads, 600 μL buffer; failed to extract DNA | |
| FastDNA® SPIN kit for soil | 0.36–1.34 μg/g of soil | 0.03–0.24/1.12–1.50 | Lysis tube E (2 mL), mechanical lysis via matrix E (1·4 mm ceramic spheres, 0·1 mm silica spheres and one 4 mm glass bead) | |
| [50] | Power Lyzer™ PowerSoil® DNA Isolation Kit | 8.7–47.5 μg/g of soil | 1.8–1.9/1.5–2.1 | - |
| the ISO standardmethod modified by Plassart et al. [51] | 21.5–43.4 μg/g of soil | 1.5/1.6–1.8 | Include mechanical lysis step; | |
| GnS-GII protocol | 8.2–49.7 μg/g of soil | 1.6–1.7/1.5–1.6 | Lysis with glass beads in different sizes and with freeze–thaw; precipitation in icecold isopropanol, humic removing by two different minicolumns | |
| Protocol A | 10 µg/mL | 1.9/2.4 | homogenization buffer consist of 100 mm Tris HCL, 100 mm EDTA, 1.5 M NaCl and 30 mg lysozyme | |
| [52] | Protocol B | 14 µg/mL | 1.6/0.65 | three-step soil washing |
| Soil DNA extraction kit, (MACHEREY-NAGEL) | 14 µg/mL | 2.2/0.86 | - | |
| Protocol D | 135 µg/mL | 2/2.2 | utilizing of physical (silica beads, freeze and melt) and chemical lysis simultaneously on both sediments (from primary stage and after homogenization buffer) | |
| [53] | PowerSoil® DNA Isolation Kit | 0–1203 ng | 2.02–2.12/0.82–1.77 | silica column-based DNA extraction with extensive washing |
| FastDNA™ SPIN Kit for Soil | 1914–20,333 ng | 1.26–1.87/0.06–0.35 | uses a large amount of binding material | |
| SDE method | 468–2913 | 1.29–1.31/0.60–0.87 | Lysis with 1 mm diameter garnet particles and three metal 4 mm bearings, removal humic acids with aluminum sulfate; gDNA purification with magnetic beads |
A260/280—absorbance ratio at 260 nm/280 nm indicated presence of protein contaminants; A260/230—absorbance ratio at 260 nm/230 nm indicated presence of humic acids; n.d.: not detectable.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wydro, U. Soil Microbiome Study Based on DNA Extraction: A Review. Water 2022, 14, 3999. https://doi.org/10.3390/w14243999
AMA Style
Wydro U. Soil Microbiome Study Based on DNA Extraction: A Review. Water. 2022; 14(24):3999. https://doi.org/10.3390/w14243999
Chicago/Turabian StyleWydro, Urszula. 2022. "Soil Microbiome Study Based on DNA Extraction: A Review" Water 14, no. 24: 3999. https://doi.org/10.3390/w14243999
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.