Bioremediation of a Polluted Groundwater: Microbial Community Comparison of Treated and Untreated Aquifer through Next Generation Sequencing

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
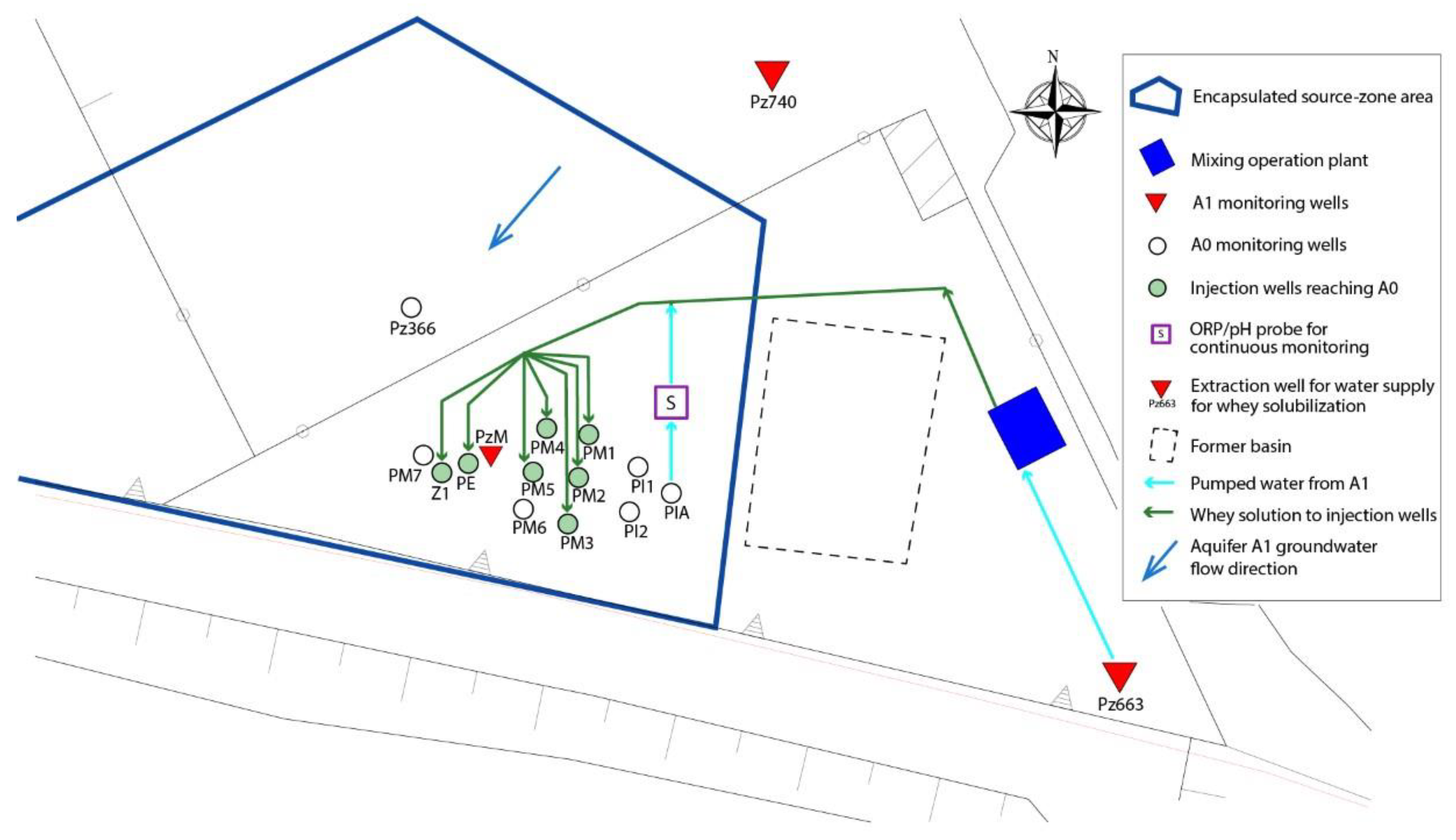
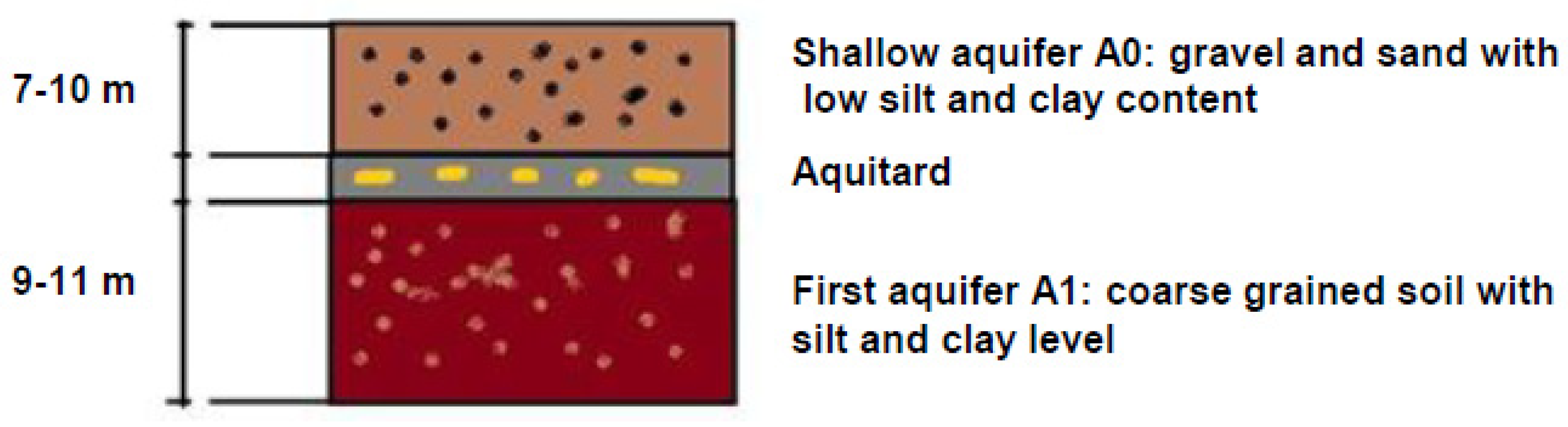
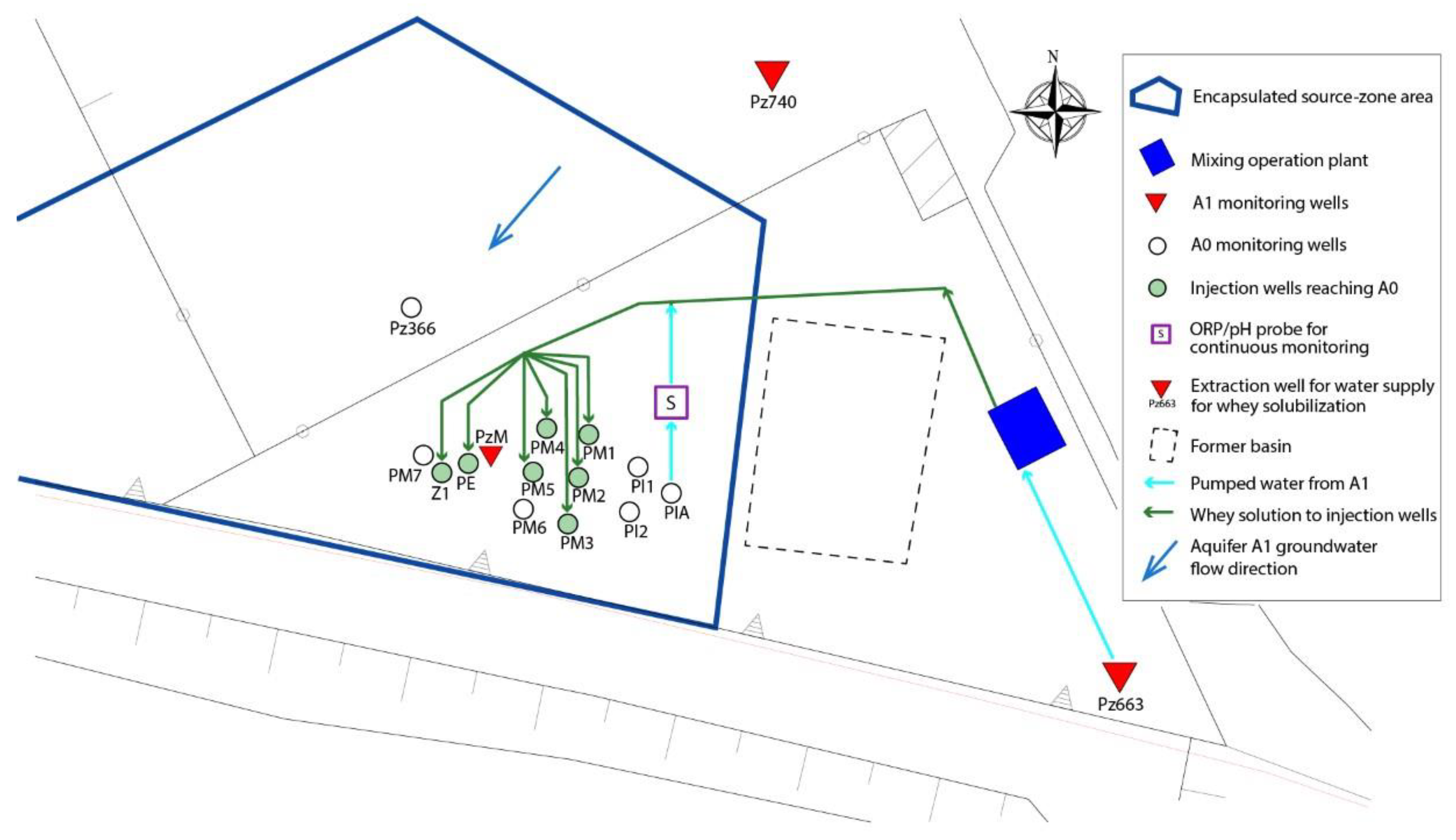
2.1. Site Characterization
2.2. In Situ Field Test: Bioremediation Implementation
2.3. Sample Collection and Total Community DNA Extraction
2.4. Sample Collection and Chemical Analysis
2.5. Next Generation Sequencing
2.6. Sequence Read Processing, Classification of Metagenomic Profiles and Further Analysis
3. Results
3.1. Contaminant Concentrations
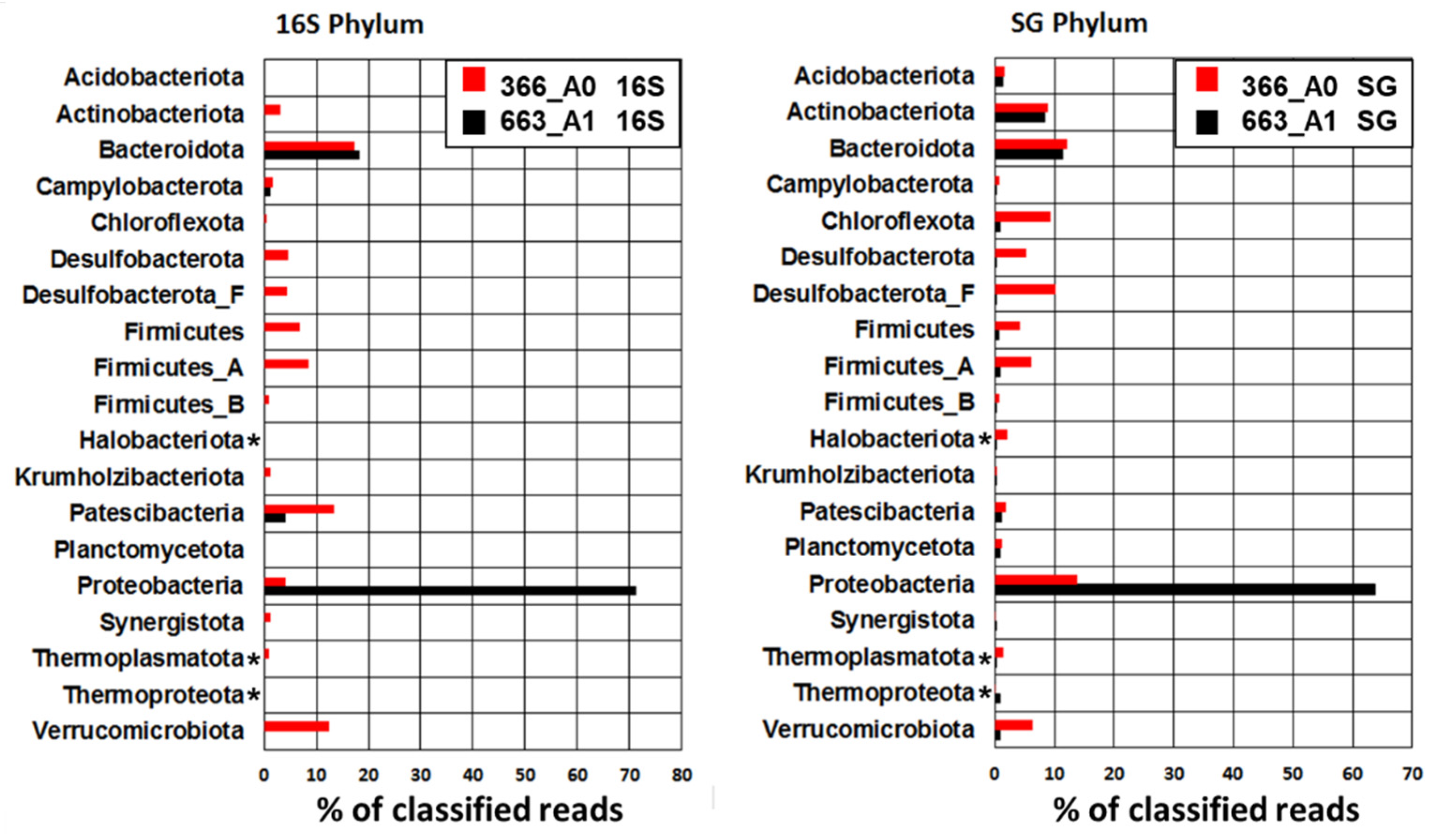
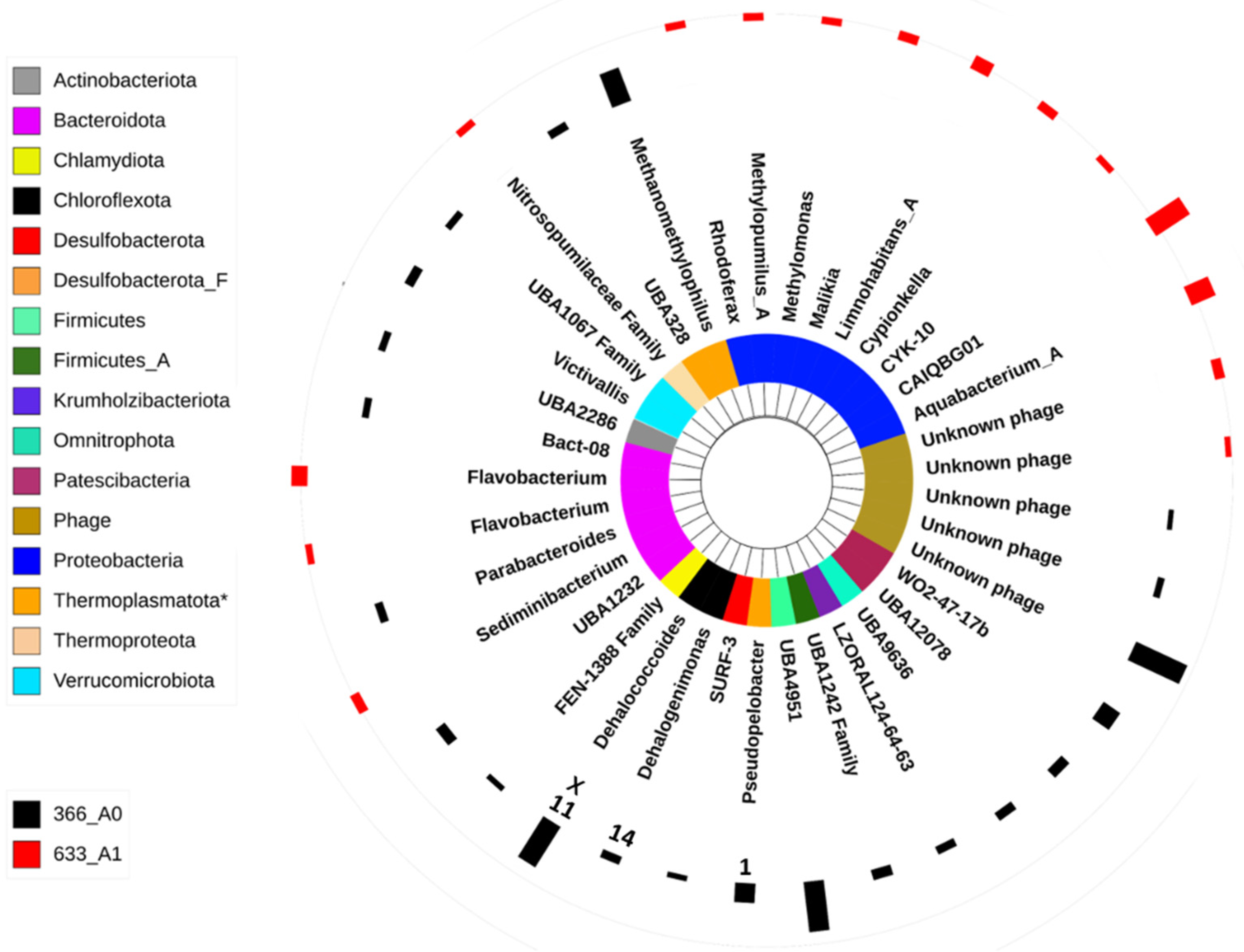
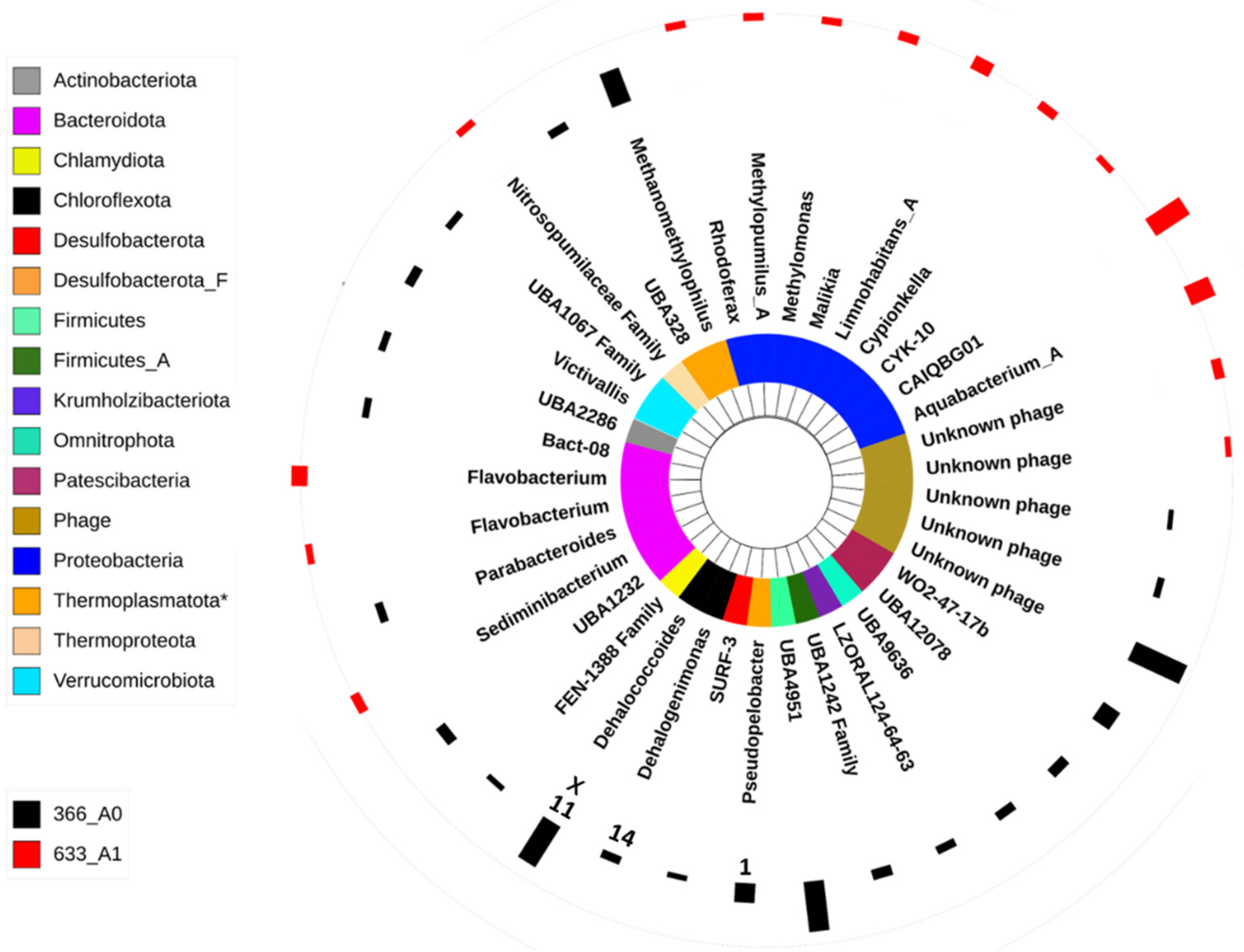
3.2. Microbial Community Composition
4. Discussion
4.1. Hydrology Data
4.2. Chemical Data: Contaminants and Fatty Acids
4.3. Microbial Composition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bajpai, P. Recycling and Deinking of Recovered Paper; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Jackson, R.E.; Dwarakanath, V. Chlorinated Decreasing Solvents: Physical-Chemical Properties Affecting Aquifer Contamination and Remediation. Groundw. Monit. Remediat. 1999, 19, 102–110. [Google Scholar] [CrossRef]
- McCarty, P.L. Groundwater Contamination by Chlorinated Solvents: History, Remediation Technologies and Strategies. In In Situ Remediation of Chlorinated Solvent Plumes; Stroo, H.F., Ward, C.H., Eds.; Springer: New York, NY, USA, 2010; pp. 1–28. [Google Scholar]
- Endo, K. Synthesis and structure of poly(vinyl chloride). Prog. Polym. Sci. 2002, 27, 2021–2054. [Google Scholar] [CrossRef]
- Matthieu, D.E., 3rd; Brusseau, M.L.; Guo, Z.; Plaschke, M.; Carroll, K.C.; Brinker, F. Persistence of a Groundwater Contaminant Plume after Hydraulic Source Containment at a Chlorinated-Solvent Contaminated Site. Ground Water Monit. Remediat. 2014, 34, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halloran, L.J.S.; Hunkeler, D. Controls on the persistence of aqueous-phase groundwater contaminants in the presence of reactive back-diffusion. Sci. Total Environ. 2020, 722, 137749. [Google Scholar] [CrossRef]
- Weigold, P.; El-Hadidi, M.; Ruecker, A.; Huson, D.H.; Scholten, T.; Jochmann, M.; Kappler, A.; Behrens, S. A metagenomic-based survey of microbial (de)halogenation potential in a German forest soil. Sci. Rep. 2016, 6, 28958. [Google Scholar] [CrossRef]
- Field, J.A.; Sierra-Alvarez, R. Biodegradability of chlorinated solvents and related chlorinated aliphatic compounds. Rev. Environ. Sci. Biol. Technol. 2004, 3, 185–254. [Google Scholar] [CrossRef]
- Vogel, T.M.; Criddle, C.S.; McCarty, P.L. ES&T Critical Reviews: Transformations of halogenated aliphatic compounds. Environ. Sci. Technol. 1987, 21, 722–736. [Google Scholar] [CrossRef]
- Scholz-Muramatsu, H.; Neumann, A.; Meßmer, M.; Moore, E.; Diekert, G. Isolation and characterization of Dehalospirillum multivorans gen. nov., sp. nov., a tetrachloroethene-utilizing, strictly anaerobic bacterium. Arch. Microbiol. 1995, 163, 48–56. [Google Scholar] [CrossRef]
- Krumholz, L.R. Desulfuromonas chloroethenica sp. nov. Uses Tetrachloroethylene and Trichloroethylene as Electron Acceptors. Int. J. Syst. Evol. Microbiol. 1997, 47, 1262–1263. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.; Fletcher, K.E.; Ritalahti, K.M.; Apkarian, R.P.; Ramos-Hernández, N.; Sanford, R.A.; Mesbah, N.M.; Löffler, F.E. Geobacter lovleyi sp. nov. strain SZ, a novel metal-reducing and tetrachloroethene-dechlorinating bacterium. Appl. Environ. Microbiol. 2006, 72, 2775–2782. [Google Scholar] [CrossRef] [Green Version]
- Löffler, F.E.; Yan, J.; Ritalahti, K.M.; Adrian, L.; Edwards, E.A.; Konstantinidis, K.T.; Müller, J.A.; Fullerton, H.; Zinder, S.H.; Spormann, A.M. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 2013, 63, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliger, C.; Schraa, G.; Stams, A.J.; Zehnder, A.J. A highly purified enrichment culture couples the reductive dechlorination of tetrachloroethene to growth. Appl. Environ. Microbiol. 1993, 59, 2991–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliger, C.; Hahn, D.; Harmsen, H.; Ludwig, W.; Schumacher, W.; Tindall, B.; Vazquez, F.; Weiss, N.; Zehnder, A.J. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra—and trichloroethene in an anaerobic respiration. Arch. Microbiol. 1998, 169, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Scholz-Muramatsu, H.; Diekert, G. Tetrachloroethene metabolism of Dehalospirillum multivorans. Arch. Microbiol. 1994, 162, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Manchester, M.J.; Hug, L.A.; Zarek, M.; Zila, A.; Edwards, E.A. Discovery of a trans-dichloroethene-respiring Dehalogenimonas species in the 1,1,2,2-tetrachloroethane-dechlorinating WBC-2 consortium. Appl. Environ. Microbiol. 2012, 78, 5280–5287. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Higgins, S.A.; Yan, J.; Şimşir, B.; Chourey, K.; Iyer, R.; Hettich, R.L.; Baldwin, B.; Ogles, D.M.; Löffler, F.E. Grape pomace compost harbors organohalide-respiring Dehalogenimonas species with novel reductive dehalogenase genes. ISME J. 2017, 11, 2767–2780. [Google Scholar] [CrossRef] [Green Version]
- Ballerstedt, H.; Hantke, J.; Bunge, M.; Werner, B.; Gerritse, J.; Andreesen, J.R.; Lechner, U. Properties of a trichlorodibenzo-p-dioxin-dechlorinating mixed culture with a Dehalococcoides as putative dechlorinating species. FEMS Microbiol. Ecol. 2004, 47, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Luijten, M.; de Weert, J.; Smidt, H.; Boschker, H.T.S.; de Vos, W.M.; Schraa, G.; Stams, A.J.M. Description of Sulfurospirillum halorespirans sp. nov., an anaerobic, tetrachloroethene-respiring bacterium, and transfer of Dehalospirillum multivorans to the genus Sulfurospirillum as Sulfurospirillum multivorans comb. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Amos, B.K.; Suchomel, E.J.; Pennell, K.D.; Löffler, F.E. Spatial and temporal distributions of Geobacter lovleyi and Dehalococcoides spp. during bioenhanced PCE-NAPL dissolution. Environ. Sci. Technol. 2009, 43, 1977–1985. [Google Scholar] [CrossRef]
- Fathepure, B.Z.; Nengu, J.P.; Boyd, S.A. Anaerobic bacteria that dechlorinate perchloroethene. Appl. Environ. Microbiol. 1987, 53, 2671–2674. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, P.E.; Ferry, J.G. Reductive dechlorination of trichloroethylene by the CO-reduced CO dehydrogenase enzyme complex from Methanosarcina thermophila. FEMS Microbiol. Lett. 1992, 75, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Beiko, R.G.; Rowe, A.R.; Richardson, R.E.; Edwards, E.A. Comparative metagenomics of three Dehalococcoides-containing enrichment cultures: The role of the non-dechlorinating community. BMC Genom. 2012, 13, 327. [Google Scholar] [CrossRef] [Green Version]
- Kotik, M.; Davidová, A.; Voříšková, J.; Baldrian, P. Bacterial communities in tetrachloroethene-polluted groundwaters: A case study. Sci. Total Environ. 2013, 454–455, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Elshahed, M.S.; McInerney, M.J. Chapter 6 Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 2009; Volume 66, pp. 141–251. [Google Scholar]
- Semkiw, E.S.; Barcelona, M.J. Field Study of Enhanced TCE Reductive Dechlorination by a Full-Scale Whey PRB. Groundw. Monit. Remediat. 2011, 31, 68–78. [Google Scholar] [CrossRef]
- Lee, P.K.H.; Warnecke, F.; Brodie, E.L.; Macbeth, T.W.; Conrad, M.E.; Andersen, G.L.; Alvarez-Cohen, L. Phylogenetic Microarray Analysis of a Microbial Community Performing Reductive Dechlorination at a TCE-Contaminated Site. Environ. Sci. Technol. 2012, 46, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Němeček, J.; Steinová, J.; Špánek, R.; Pluhař, T.; Pokorný, P.; Najmanová, P.; Knytl, V.; Černík, M. Thermally enhanced in situ bioremediation of groundwater contaminated with chlorinated solvents—A field test. Sci. Total Environ. 2018, 622–623, 743–755. [Google Scholar] [CrossRef]
- Aulenta, F.; Canosa, A.; Leccese, M.; Petrangeli Papini, M.; Majone, M.; Viotti, P. Field Study of In Situ Anaerobic Bioremediation of a Chlorinated Solvent Source Zone. Ind. Eng. Chem. Res. 2007, 46, 6812–6819. [Google Scholar] [CrossRef]
- Stroo, H.F.; West, M.R.; Kueper, B.H.; Borden, R.C.; Major, D.W.; Ward, C.H. In Situ Bioremediation of Chlorinated Ethene: DNAPL Source Zones; ITRC, Interstate Technology & Regulatory Council: Washington, DC, USA, 2008. [Google Scholar]
- Hinlo, R.; Gleeson, D.; Lintermans, M.; Furlan, E. Methods to maximise recovery of environmental DNA from water samples. PLoS ONE 2017, 12, e0179251. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 2016, 074161. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitua, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; McGuinness, K.A.; Ophori, D.U. A field evaluation of enhanced reductive dechlorination of chlorinated solvents in groundwater, New York Metropolitan Area. Environ. Geol. 2004, 45, 623–632. [Google Scholar] [CrossRef]
- Smith, S.; Dupont, R.R.; McLean, J.E. Arsenic Release and Attenuation Processes in a Groundwater Aquifer During Anaerobic Remediation of TCE with Biostimulation. Groundw. Monit. Remediat. 2019, 39, 61–70. [Google Scholar] [CrossRef]
- Macbeth, T.W.; Nelson, L.; Rothermel, J.S.; Wymore, R.A.; Sorenson, K.S. Evaluation of Whey for Bioremediation of Trichloroethene Source Zones. Bioremediation J. 2006, 10, 115–128. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Futamata, H.; Yoshida, N.; Kurogi, T.; Kaiya, S.; Hiraishi, A. Reductive dechlorination of chloroethenes by Dehalococcoides-containing cultures enriched from a polychlorinated-dioxin-contaminated microcosm. ISME J. 2007, 1, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Borrel, G.; Parisot, N.; Harris, H.M.B.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genom. 2014, 15, 679. [Google Scholar] [CrossRef] [Green Version]
- Duhamel, M.; Edwards, E.A. Microbial composition of chlorinated ethene-degrading cultures dominated by Dehalococcoides. FEMS Microbiol. Ecol. 2006, 58, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.R.; Lazar, B.J.; Morris, R.M.; Richardson, R.E. Characterization of the community structure of a dechlorinating mixed culture and comparisons of gene expression in planktonic and biofloc-associated “Dehalococcoides” and Methanospirillum species. Appl. Environ. Microbiol. 2008, 74, 6709–6719. [Google Scholar] [CrossRef] [Green Version]
- Moe, W.M.; Yan, J.; Nobre, M.F.; da Costa, M.S.; Rainey, F.A. Dehalogenimonas lykanthroporepellens gen. nov., sp. nov., a reductive dehalogenating bacterium isolated from chlorinated solvent contaminated groundwater. Int. J. Syst. Evol. Microbiol. 2009, 59, 2692–2697. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Masuda, Y.; Wang, X.; Ushijima, N.; Shiratori, Y.; Senoo, K.; Itoh, H. Genome-Based Taxonomic Rearrangement of the Order Geobacterales Including the Description of Geomonas azotofigens sp. nov. and Geomonas diazotrophica sp. nov. Front. Microbiol. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Mattes, T.; Oh Jin, Y.; Livermore, J.; Pearl, M.; Liu, X. Abundance and activity of vinyl chloride (VC)-oxidizing bacteria in a dilute groundwater VC plume biostimulated with oxygen and ethene. Appl. Microbiol. Biotechnol. 2015, 99, 9267–9276. [Google Scholar] [CrossRef] [PubMed]
- Maness, A.D.; Bowman, K.S.; Yan, J.; Rainey, F.A.; Moe, W.M. Dehalogenimonas spp. can Reductively Dehalogenate High Concentrations of 1,2-Dichloroethane, 1,2-Dichloropropane, and 1,1,2-Trichloroethane. AMB Express 2012, 2, 54. [Google Scholar] [CrossRef] [Green Version]
- Lang, K.; Schuldes, J.; Klingl, A.; Poehlein, A.; Daniel, R.; Brunea, A. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus methanoplasma termitum”. Appl. Environ. Microbiol. 2015, 81, 1338–1352. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Heal, K.R.; Ingalls, A.E.; Doxey, A.C.; Neufeld, J.D. Metagenomic and chemical characterization of soil cobalamin production. ISME J. 2020, 14, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Huling, S.G.; Weaver, J.W. Ground Water Issue. Dense Nonaqueous Phase Liquids; EPA: Washington, DC, USA, 1991. [Google Scholar]
- Ayral-Çınar, D.; Demond, A.H. Accumulation of DNAPL waste in subsurface clayey lenses and layers. J. Contam. Hydrol. 2020, 229, 103579. [Google Scholar] [CrossRef]
- Sale, T.C.; Zimbron, J.A.; Dandy, D.S. Effects of reduced contaminant loading on downgradient water quality in an idealized two-layer granular porous media. J. Contam. Hydrol. 2008, 102, 72–85. [Google Scholar] [CrossRef]
- Falta, R.W.; Suresh Rao, P.; Basu, N. Assessing the impacts of partial mass depletion in DNAPL source zones: I. Analytical modeling of source strength functions and plume response. J. Contam. Hydrol. 2005, 78, 259–280. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, M.; Frascari, D.; Pinelli, D.; Mezzetti, F.; Fedi, S.; Zannoni, D. Aerobic cometabolism of 1,1,2,2-TeCA by a propane-growing microbial consortium (C2): Diversity of alkane monooxygenase genes and design of an on-site bioremediation process. Int. Biodeterior. Biodegrad. 2017, 119, 649–660. [Google Scholar] [CrossRef]
- Fischer, M.A.; Güllert, S.; Neulinger, S.C.; Streit, W.R.; Schmitz, R.A. Evaluation of 16S rRNA Gene Primer Pairs for Monitoring Microbial Community Structures Showed High Reproducibility within and Low Comparability between Datasets Generated with Multiple Archaeal and Bacterial Primer Pairs. Front. Microbiol. 2016, 7, 1297. [Google Scholar] [CrossRef] [PubMed]
- Sambo, F.; Finotello, F.; Lavezzo, E.; Baruzzo, G.; Masi, G.; Peta, E.; Falda, M.; Toppo, S.; Barzon, L.; Di Camillo, B. Optimizing PCR primers targeting the bacterial 16S ribosomal RNA gene. BMC Bioinform. 2018, 19, 343. [Google Scholar] [CrossRef] [PubMed]
- Chau, A.T.T.; Lee, M.; Adrian, L.; Manefield, M.J. Syntrophic Partners Enhance Growth and Respiratory Dehalogenation of Hexachlorobenzene by Dehalococcoides mccartyi Strain CBDB1. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Im, J.; Yang, Y.; Löffler, F.E. Guided cobalamin biosynthesis supports Dehalococcoides mccartyi reductive dechlorination activity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120320. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, C.; Zhao, S.; He, J. Microbial synergistic interactions for reductive dechlorination of polychlorinated biphenyls. Sci. Total Environ. 2019, 666, 368–376. [Google Scholar] [CrossRef]
- Nagymáté, Z.; Jurecska, L.; Romsics, C.; Tóth, F.; Bódai, V.; Mészáros, É.; Szabó, A.; Erdélyi, B.; Márialigeti, K. Preparation and characterization of site-specific dechlorinating microbial inocula capable of complete dechlorination enriched in anaerobic microcosms amended with clay mineral. World J. Microbiol. Biotechnol. 2020, 36, 29. [Google Scholar] [CrossRef] [Green Version]
- Kengen, S.W.; Breidenbach, C.G.; Felske, A.; Stams, A.J.; Schraa, G.; de Vos, W.M. Reductive dechlorination of tetrachloroethene to cis-1, 2-dichloroethene by a thermophilic anaerobic enrichment culture. Appl. Environ. Microbiol. 1999, 65, 2312–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezberová, J.; Jezbera, J.; Brandt, U.; Lindström, E.S.; Langenheder, S.; Hahn, M.W. Ubiquity of Polynucleobacter necessarius ssp. asymbioticus in lentic freshwater habitats of a heterogeneous 2000 km area. Environ. Microbiol. 2010, 12, 658–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arp, D.J.; Yeager, C.M.; Hyman, M.R. Molecular and cellular fundamentals of aerobic cometabolism of trichloroethylene. Biodegradation 2001, 12, 81–103. [Google Scholar] [CrossRef]
- Chee, G.J. Biodegradation analyses of trichloroethylene (TCE) by bacteria and its use for biosensing of TCE. Talanta 2011, 85, 1778–1782. [Google Scholar] [CrossRef]
- Ding, L.; Yokota, A. Curvibacter fontana sp. nov., a microaerobic bacteria isolated from well water. J. Gen. Appl. Microbiol. 2010, 56, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.K.; Yang, H.Y.; Huang, S.R.; Hung, J.M.; Lu, C.J.; Liu, M.H. Complete degradation of chlorinated ethenes and its intermediates through sequential anaerobic/aerobic biodegradation in simulated groundwater columns (complete degradation of chlorinated ethenes). Int. J. Environ. Sci. Technol. 2020, 17, 4517–4530. [Google Scholar] [CrossRef]
- Jin, D.; Zhang, F.; Shi, Y.; Kong, X.; Xie, Y.; Du, X.; Li, Y.; Zhang, R. Diversity of bacteria and archaea in the groundwater contaminated by chlorinated solvents undergoing natural attenuation. Environ. Res. 2020, 185, 109457. [Google Scholar] [CrossRef]
- Satsuma, K.; Masuda, M.; Sato, K. O-Demethylation and successive oxidative dechlorination of methoxychlor by Bradyrhizobium sp. strain 17-4, isolated from river sediment. Appl. Environ. Microbiol. 2012, 78, 5313–5319. [Google Scholar] [CrossRef] [Green Version]
- Newman, L.M.; Wackett, L.P. Fate of 2,2,2-trichloroacetaldehyde (chloral hydrate) produced during trichloroethylene oxidation by methanotrophs. Appl. Environ. Microbiol. 1991, 57, 2399–2402. [Google Scholar] [CrossRef] [Green Version]
- Tsien, H.C.; Brusseau, G.A.; Hanson, R.S.; Waclett, L.P. Biodegradation of trichloroethylene by Methylosinus trichosporium OB3b. Appl. Environ. Microbiol. 1989, 55, 3155–3161. [Google Scholar] [CrossRef] [Green Version]
- Fogel, M.M.; Taddeo, A.R.; Fogel, S. Biodegradation of chlorinated ethenes by a methane-utilizing mixed culture. Appl. Environ. Microbiol. 1986, 51, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pz366 (A0) | Pz663 (A1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Date | 2 February 2017 | 31 May 2017 | 12 July 2017 | 12 September 2017 | 8 May 2018 | 20 February 2017 | 31 May 2017 | 12 July 2017 | 12 September 2017 | 8 May 2018 |
| 1,1,2,2 TeCA | 1400 | 780 | 240 | 140 | 1860 | 70 | 37 | 84 | 53 | 36 |
| TCE | 520 | 110 | 17 | 19 | 25.2 | 54 | 96 | 110 | 23 | 78 |
| PCE | 96 | 4.2 | 6.5 | 3.7 | 6.4 | 4.6 | 4.9 | 11 | 6.0 | 5.2 |
| cis-DCE | 10 | 450 | 520 | 610 | 142 | 0.22 | 1.1 | 15 | 2.9 | 3.11 |
| trans-DCE | 3.5 | 240 | 410 | 480 | 330 | 0 | <2.0 | 0.66 | 1.2 | 0.196 |
| 1,1,2 TCA | 8.8 | 120 | 41 | 31 | 71 | 0 | <2.0 | 1.1 | 0.84 | 0.25 |
| VC | 1.1 | 120 | 220 | 120 | 176 | <0.025 | <2.5 | 0.28 | 0.43 | 0.103 |
| ETH | n.d. | n.d. | n.d. | 68 | <5 | n.d. | n.d. | n.d. | <7.4 | <5 |
| METH | n.d. | n.d. | n.d. | 410 | 4500 | n.d. | n.d. | n.d. | <3.8 | <5 |
| Sample | Number of Reads | Number after QC | Phylum % | Class % | Order % | Family % | Genus % | Species % |
|---|---|---|---|---|---|---|---|---|
| 663 (A1)16S | 62,551 | 41,569 * | 95.8 | 94.8 | 89.7 | 81.1 | 49.1 | 8.3 |
| 366 (A0) 16S | 63,906 | 42,128 * | 83.3 | 69.1 | 61.1 | 44.2 | 28.0 | 15.5 |
| 663 (A1) SG | 20,825,527 | 14,757,073 | 52.46 | 51.69 | 49.71 | 47.86 | 39.20 | 23.19 |
| 366 (A0) SG | 18,240,703 | 11,635,396 | 38.27 | 37.74 | 36.35 | 34.44 | 26.72 | 16.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pretto, P.; Sanseverino, I.; Demichelis, F.; Lotti, F.; Lahm, A.; Garcia Perez, A.; Ricci, R.; Lettieri, T. Bioremediation of a Polluted Groundwater: Microbial Community Comparison of Treated and Untreated Aquifer through Next Generation Sequencing. Water 2022, 14, 2456. https://doi.org/10.3390/w14162456
Pretto P, Sanseverino I, Demichelis F, Lotti F, Lahm A, Garcia Perez A, Ricci R, Lettieri T. Bioremediation of a Polluted Groundwater: Microbial Community Comparison of Treated and Untreated Aquifer through Next Generation Sequencing. Water. 2022; 14(16):2456. https://doi.org/10.3390/w14162456
Chicago/Turabian StylePretto, Patrizia, Isabella Sanseverino, Francesca Demichelis, Francesca Lotti, Armin Lahm, Angela Garcia Perez, Roberto Ricci, and Teresa Lettieri. 2022. "Bioremediation of a Polluted Groundwater: Microbial Community Comparison of Treated and Untreated Aquifer through Next Generation Sequencing" Water 14, no. 16: 2456. https://doi.org/10.3390/w14162456