
Effect of Eutrophication Control Methods on the Generation of Greenhouse Carbon Gases in Sediment



Abstract
:1. Introduction
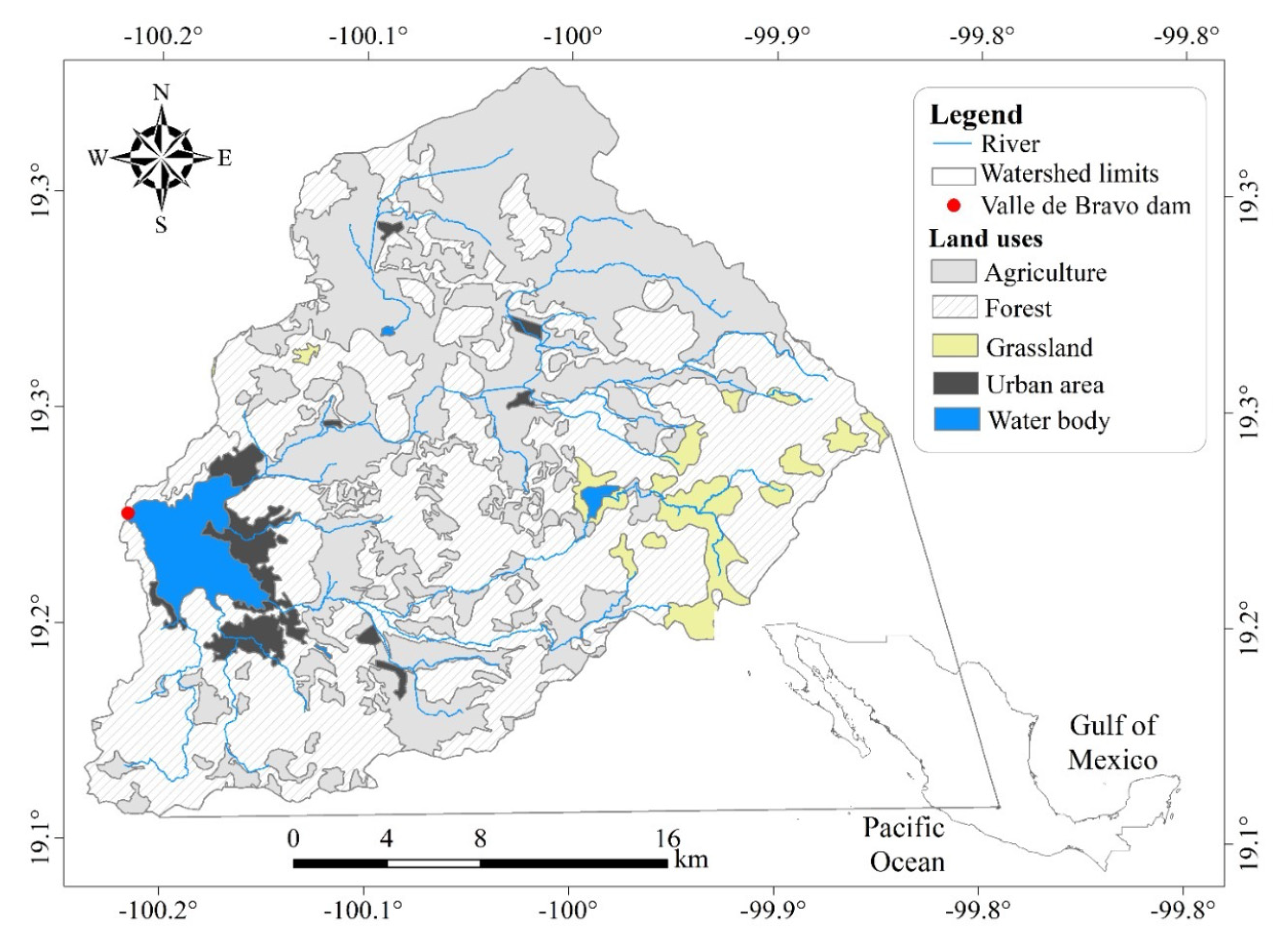
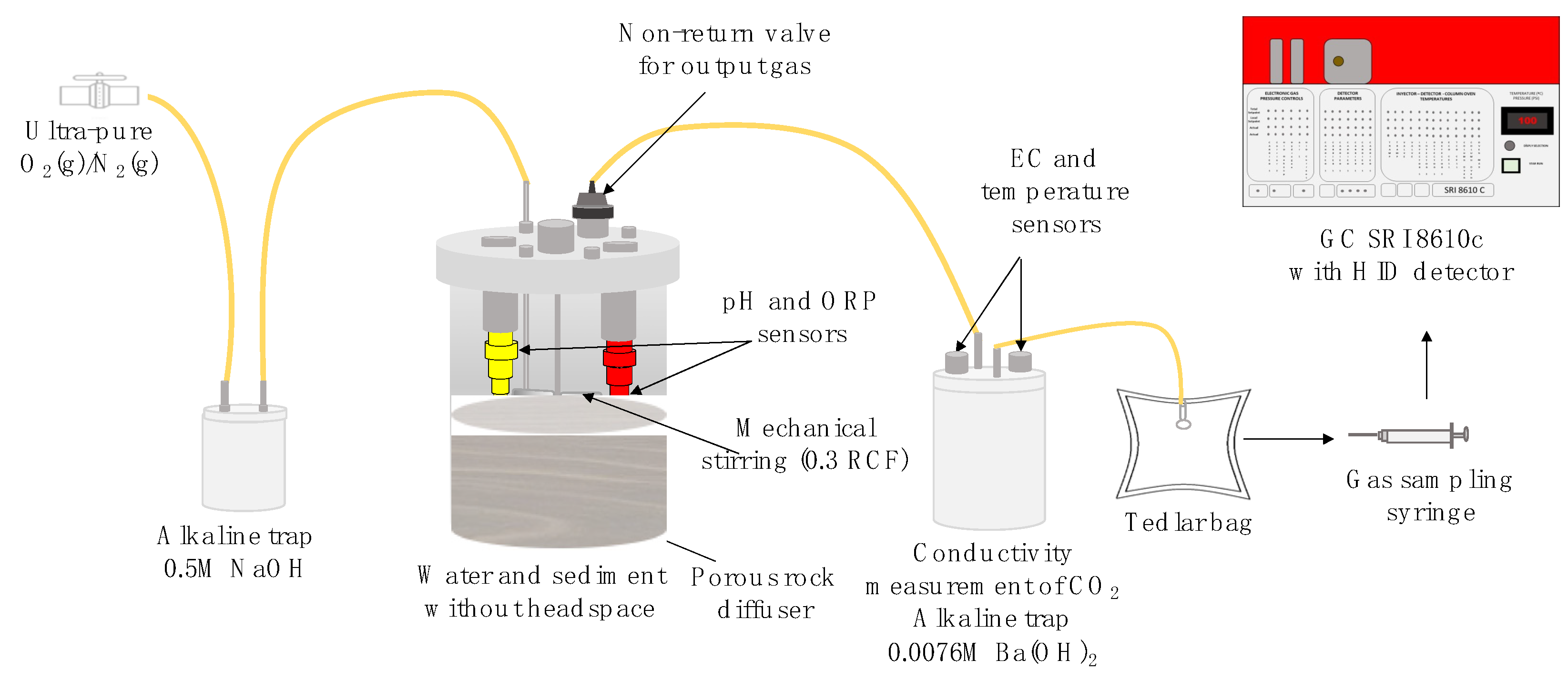
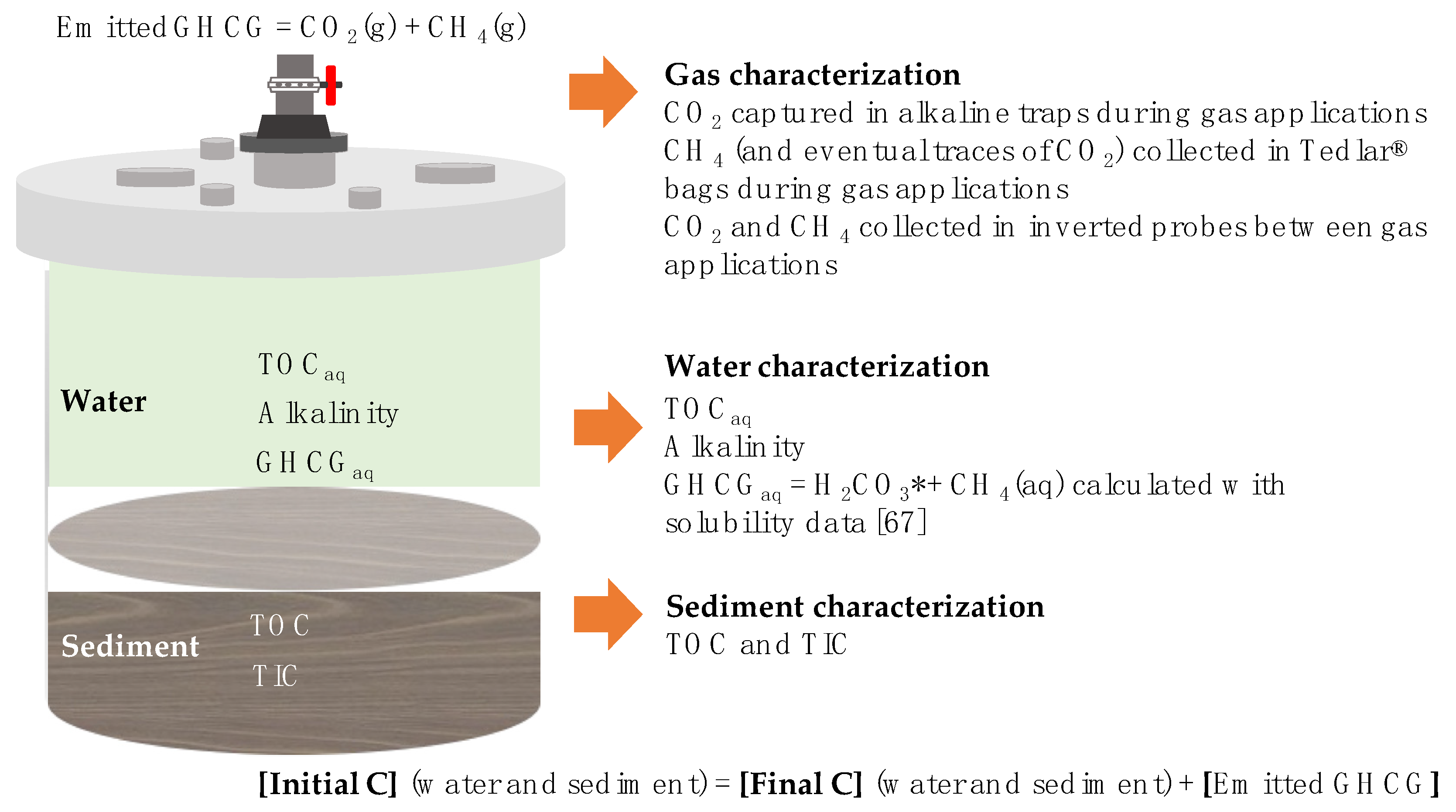
2. Materials and Methods
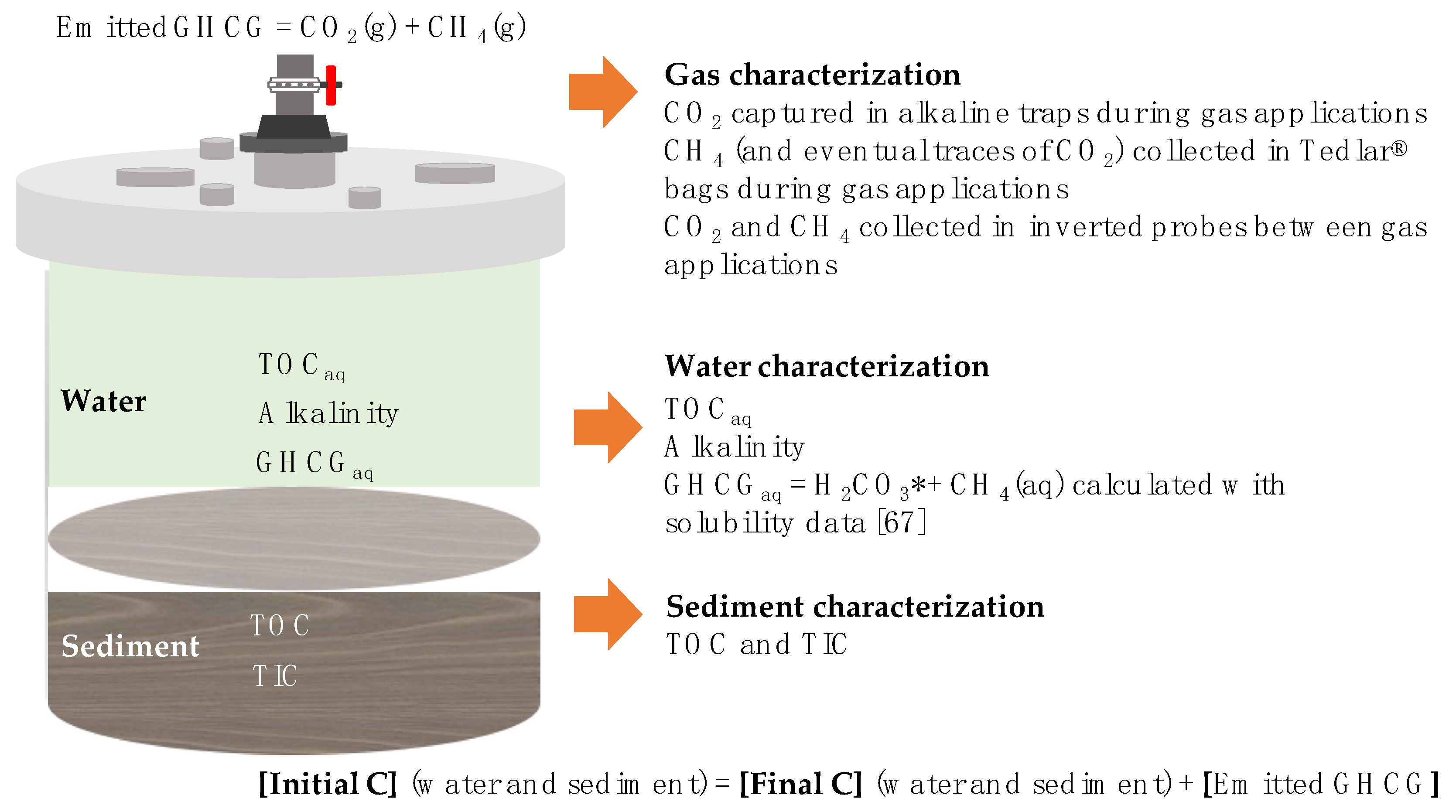
- CO2 and CH4 collected in inverted probes
- CO2 captured in alkaline traps
- CH4 (and eventual traces of CO2) collected in Tedlar bags
3. Results and Discussion
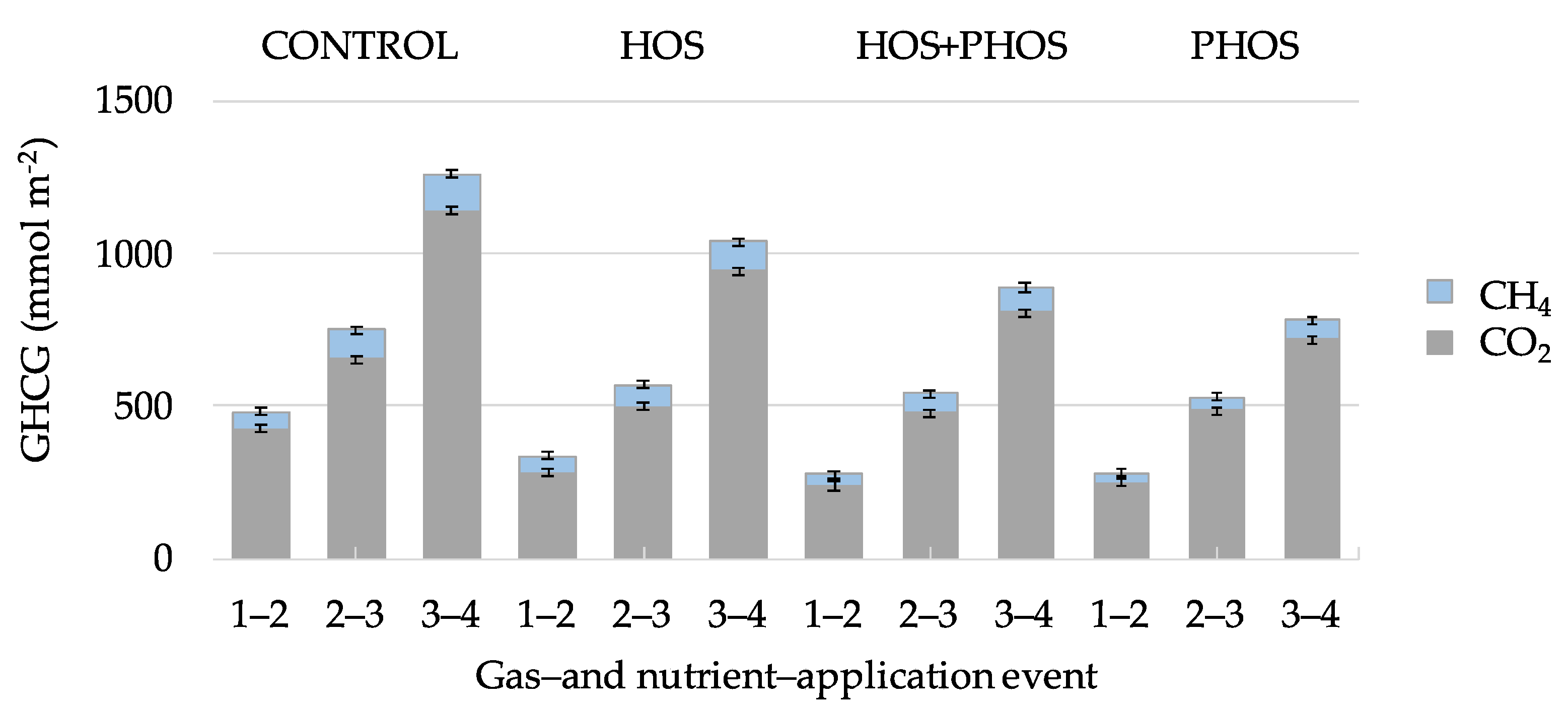
3.1. Carbon Mass-Balances
3.2. Variations in Eh and pH
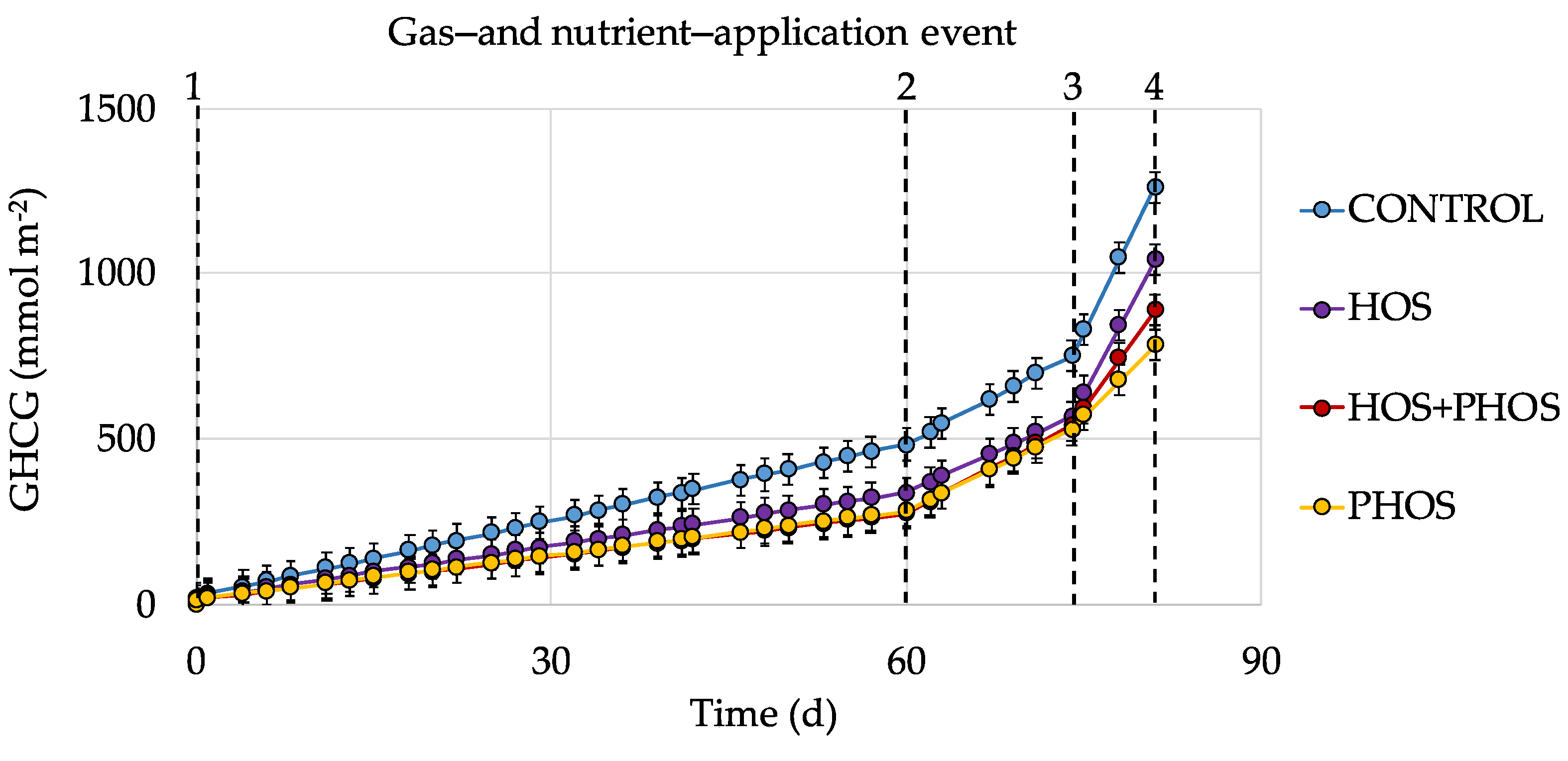
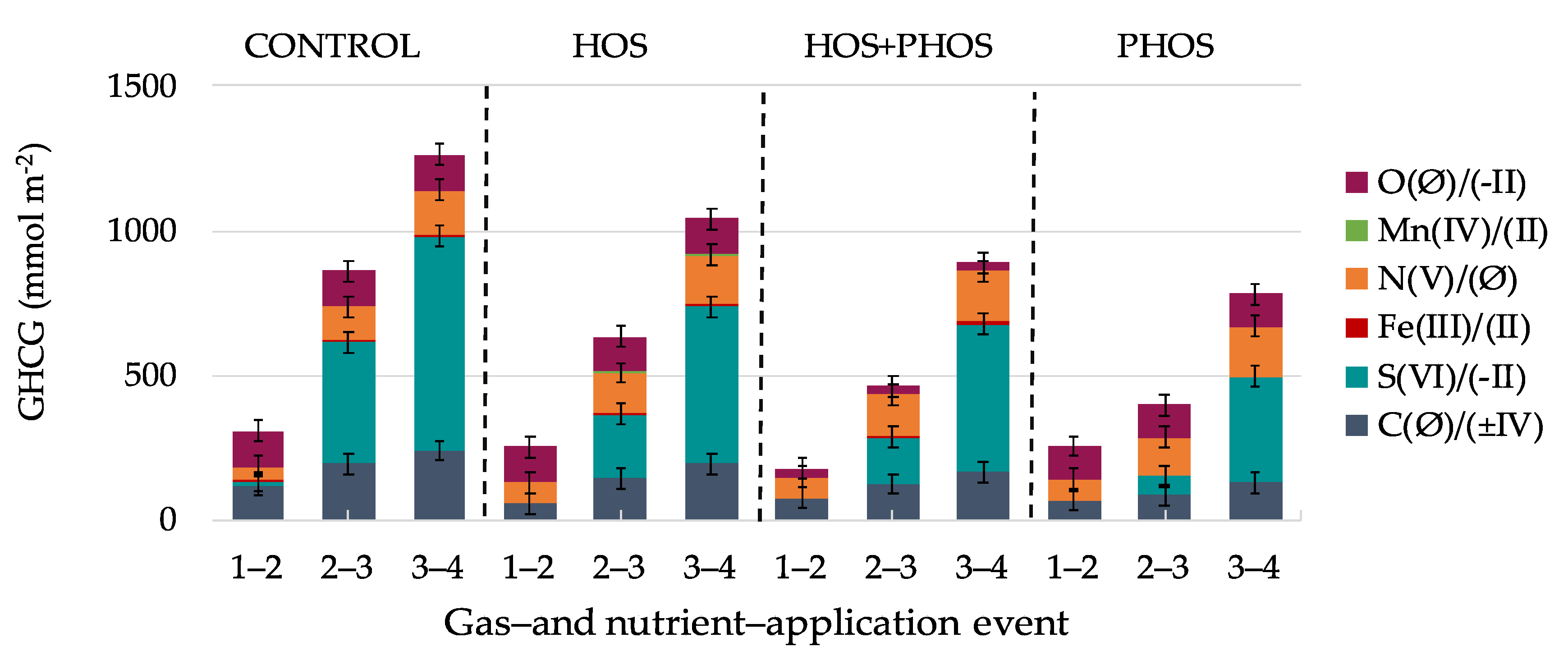
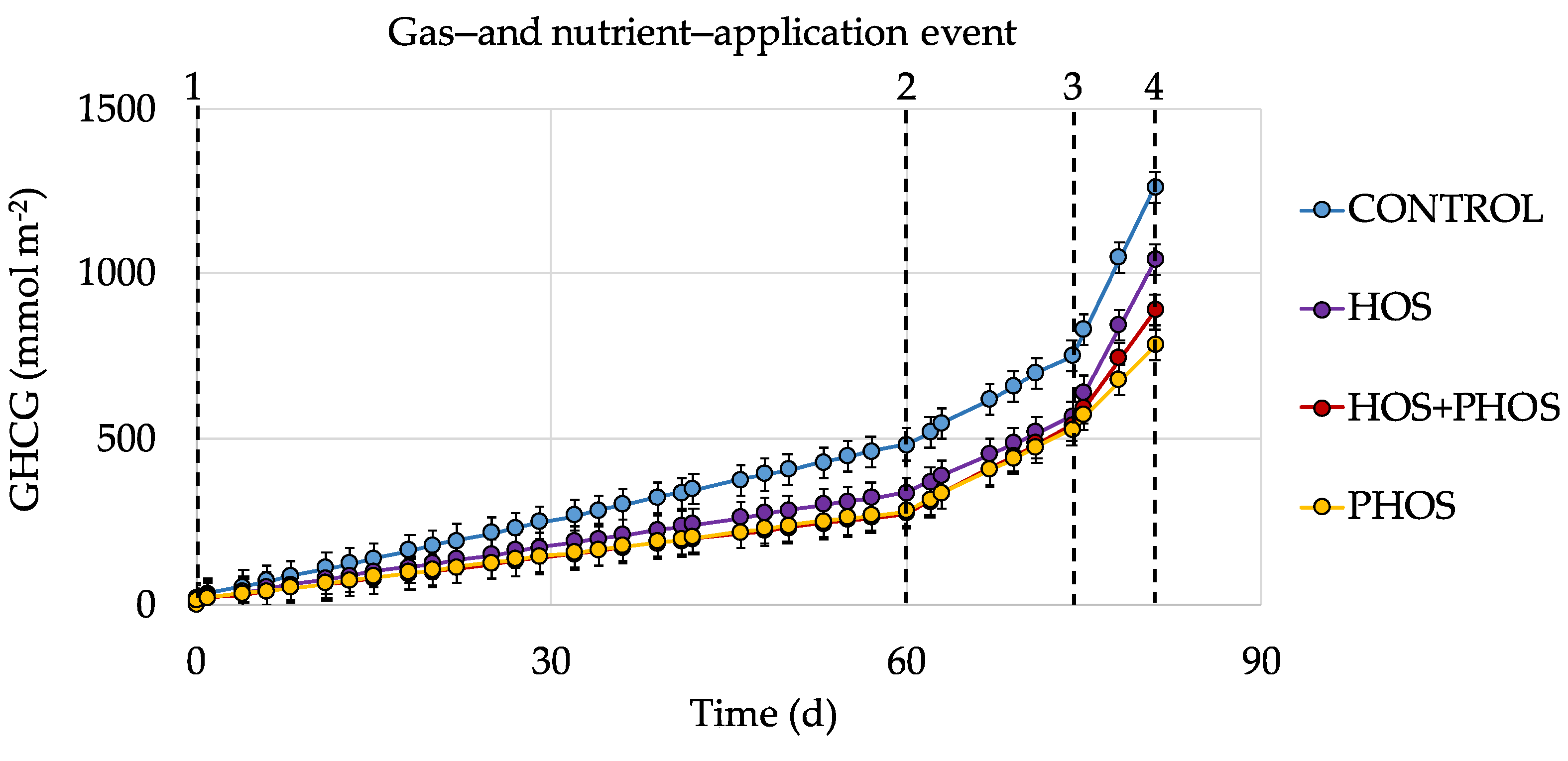
3.3. Generation Velocities of GHCG
3.4. Organic Matter Mineralization Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, V.H.; Joye, S.B.; Howarth, R.W. Eutrophication of Freshwater and Marine Ecosystems. Limnol. Oceanogr. 2006, 51, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.H.; Schindler, D.W. Eutrophication Science: Where Do We Go from Here? Trends Ecol. Evol. 2009, 24, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Raven, J.A. Aquatic Photosynthesis, 2nd ed.; Princeton University Press: Princeton, NJ, USA, 2007; ISBN 978-1-4008-4972-7. [Google Scholar]
- Burgin, A.J.; Hamilton, S.K. Have We Overemphasized the Role of Denitrification in Aquatic Ecosystems? A Review of Nitrate Removal Pathways. Front. Ecol. Environ. 2007, 5, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Thorbergsdóttir, I.M.; Reynir Gíslason, S.; Ingvason, H.R.; Einarsson, Á. Benthic Oxygen Flux in the Highly Productive Subarctic Lake Myvatn, Iceland: In Situ Benthic Flux Chamber Study. Aquat. Ecol. 2004, 38, 177–189. [Google Scholar] [CrossRef]
- Müller, B.; Bryant, L.D.; Matzinger, A.; Wüest, A. Hypolimnetic Oxygen Depletion in Eutrophic Lakes. Environ. Sci. Technol. 2012, 46, 9964–9971. [Google Scholar] [CrossRef]
- Petsch, S.T. The Global Oxygen Cycle. In Treatise on Geochemistry; Elsevier: Amsterdam, The Netherlands, 2014; pp. 437–473. ISBN 978-0-08-098300-4. [Google Scholar]
- Luu, Y.-S. Review: Microbial Mechanisms of Accessing Insoluble Fe(III) as an Energy Source. World J. Microbiol. Biotechnol. 2003, 19, 215–225. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The Biogeochemical Sulfur Cycle of Marine Sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Ouddane, B.; Boust, D.; Martin, E.; Fischer, J.C.; Wartel, M. The Post-Depositional Reactivity of Iron and Manganese in the Sediments of a Macrotidal Estuarine System. Estuaries 2001, 24, 1015. [Google Scholar] [CrossRef]
- Himmelheber, D.W.; Taillefert, M.; Pennell, K.D.; Hughes, J.B. Spatial and Temporal Evolution of Biogeochemical Processes Following In Situ Capping of Contaminated Sediments. Environ. Sci. Technol. 2008, 42, 4113–4120. [Google Scholar] [CrossRef]
- Song, Y.; Müller, G. Sediment-Water Interactions in Anoxic Freshwater Sediments; Lecture Notes in Earth Sciences; Springer: Berlin/Heidelberg, Germany, 1999; Volume 81, ISBN 978-3-540-65022-5. [Google Scholar]
- Lovley, D.R. Organic Matter Mineralization with the Reduction of Ferric Iron: A Review. Geomicrobiol. J. 1987, 5, 375–399. [Google Scholar] [CrossRef]
- Holmer, M.; Storkholm, P. Sulphate Reduction and Sulphur Cycling in Lake Sediments: A Review: Sulphate Cycling in Lake Sediments. Freshw. Biol. 2001, 46, 431–451. [Google Scholar] [CrossRef]
- Borrel, G.; Jézéquel, D.; Biderre-Petit, C.; Morel-Desrosiers, N.; Morel, J.-P.; Peyret, P.; Fonty, G.; Lehours, A.-C. Production and Consumption of Methane in Freshwater Lake Ecosystems. Res. Microbiol. 2011, 162, 832–847. [Google Scholar] [CrossRef]
- Wilson, R.M.; Tfaily, M.M.; Rich, V.I.; Keller, J.K.; Bridgham, S.D.; Zalman, C.M.; Meredith, L.; Hanson, P.J.; Hines, M.; Pfeifer-Meister, L.; et al. Hydrogenation of Organic Matter as a Terminal Electron Sink Sustains High CO2:CH4 Production Ratios during Anaerobic Decomposition. Org. Geochem. 2017, 112, 22–32. [Google Scholar] [CrossRef] [Green Version]
- St. Louis, V.L.; Kelly, C.A.; Duchemin, É.; Rudd, J.W.M.; Rosenberg, D.M. Reservoir Surfaces as Sources of Greenhouse Gases to the Atmosphere: A Global Estimate. BioScience 2000, 50, 766. [Google Scholar] [CrossRef]
- Malyan, S.K.; Singh, O.; Kumar, A.; Anand, G.; Singh, R.; Singh, S.; Yu, Z.; Kumar, J.; Fagodiya, R.K.; Kumar, A. Greenhouse Gases Trade-Off from Ponds: An Overview of Emission Process and Their Driving Factors. Water 2022, 14, 970. [Google Scholar] [CrossRef]
- Rochette, P.; Eriksen-Hamel, N.S. Chamber Measurements of Soil Nitrous Oxide Flux: Are Absolute Values Reliable? Soil Sci. Soc. Am. J. 2008, 72, 331–342. [Google Scholar] [CrossRef]
- Hansen, J.; Thamdrup, B.; Jørgensen, B. Anoxic Incubation of Sediment in Gas-Tight Plastic Bags: A Method for Biogeochemical Process Studies. Mar. Ecol. Prog. Ser. 2000, 208, 273–282. [Google Scholar] [CrossRef]
- Van Afferden, M.; Hansen, A.M.; Kaiser, C.; Chapelain, N. Laboratory Test System to Measure Microbial Respiration Rate. Int. J. Environ. Pollut. 2006, 26, 220. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, X.; Huang, P. Estimating Carbon Dioxide (CO2) Emissions from Reservoirs Using Artificial Neural Networks. Water 2018, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Seibert, S.L.; Greskowiak, J.; Prommer, H.; Böttcher, M.E.; Massmann, G. Modeling of Biogeochemical Processes in a Barrier Island Freshwater Lens (Spiekeroog, Germany). J. Hydrol. 2019, 575, 1133–1144. [Google Scholar] [CrossRef]
- Sánchez-Carrillo, S.; Alcocer, J.; Vargas-Sánchez, M.; Soria-Reinoso, I.; Rivera-Herrera, E.M.; Cortés-Guzmán, D.; Cuevas-Lara, D.; Guzmán-Arias, A.P.; Merino-Ibarra, M.; Oseguera, L.A. Greenhouse Gas Emissions from Mexican Inland Waters: First Estimation and Uncertainty Using an Upscaling Approach. Inland Waters 2022, 1–17. [Google Scholar] [CrossRef]
- SINA (Sistema Nacional de Información del Agua). Sistema Cutzamala. Available online: http://sina.conagua.gob.mx/sina/index.php (accessed on 11 August 2021).
- INEGI (Instituto Nacional de Estadística y Geografía). Resultados Generales del Censo de Población y Vivienda. 2020. Available online: https://www.inegi.org.mx/programas/ccpv/2020/ (accessed on 5 August 2021).
- Márquez-Pacheco, H.; Hansen, A.M.; Falcón-Rojas, A. Phosphorous Control in a Eutrophied Reservoir. Environ. Sci. Pollut. Res. 2013, 20, 8446–8456. [Google Scholar] [CrossRef] [PubMed]
- Nandini, S.; Sánchez-Zamora, C.; Sarma, S.S.S. Toxicity of Cyanobacterial Blooms from the Reservoir Valle de Bravo (Mexico): A Case Study on the Rotifer Brachionus Calyciflorus. Sci. Total Environ. 2019, 688, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- World Bank; CONAGUA (Comisión Nacional del Agua). Diagnóstico Para El Manejo Integral de Las Subcuencas Tuxpan, El Bosque, Ixtapan del Oro, Valle de Bravo, Colorines-Chilesdo y Villa Victoria Pertenecientes al Sistema Cutzamala. Available online: https://www.gob.mx/conagua/documentos/diagnostico-para-el-manejo-integral-de-las-subcuencas-tuxpan-el-bosque-ixtapan-del-oro-valle-de-bravo-colorines-chilesdo-y-villa-victoria-pertenecientes-al-sistema-cutzamala (accessed on 1 May 2022).
- INEGI (Instituto Nacional de Estadística y Geografía). Uso de Suelo y Vegetación, Serie V, Escala 1:250,000. Available online: https://www.inegi.org.mx/temas/usosuelo/ (accessed on 5 August 2021).
- Bishop, W.M.; Richardson, R.J. Influence of Phoslock® on Legacy Phosphorus, Nutrient Ratios, and Algal Assemblage Composition in Hypereutrophic Water Resources. Environ. Sci. Pollut. Res. 2018, 25, 4544–4557. [Google Scholar] [CrossRef] [PubMed]
- Lürling, M.; Faassen, E.J. Controlling Toxic Cyanobacteria: Effects of Dredging and Phosphorus-Binding Clay on Cyanobacteria and Microcystins. Water Res. 2012, 46, 1447–1459. [Google Scholar] [CrossRef]
- Gantzer, P.A.; Bryant, L.D.; Little, J.C. Effect of Hypolimnetic Oxygenation on Oxygen Depletion Rates in Two Water-Supply Reservoirs. Water Res. 2009, 43, 1700–1710. [Google Scholar] [CrossRef]
- Beutel, M.W.; Horne, A.J. A Review of the Effects of Hypolimnetic Oxygenation on Lake and Reservoir Water Quality. Lake Reserv. Manag. 1999, 15, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Epe, T.S.; Finsterle, K.; Yasseri, S. Nine Years of Phosphorus Management with Lanthanum Modified Bentonite (Phoslock) in a Eutrophic, Shallow Swimming Lake in Germany. Lake Reserv. Manag. 2017, 33, 119–129. [Google Scholar] [CrossRef]
- Elser, J.; Kyle, M.; Learned, J.; McCrackin, M.; Peace, A.; Steger, L. Life on the Stoichiometric Knife-Edge: Effects of High and Low Food C:P Ratio on Growth, Feeding, and Respiration in Three Daphnia Species. Inland Waters 2016, 6, 136–146. [Google Scholar] [CrossRef]
- Zamparas, M.; Zacharias, I. Restoration of Eutrophic Freshwater by Managing Internal Nutrient Loads. A Review. Sci. Total Environ. 2014, 496, 551–562. [Google Scholar] [CrossRef]
- Søndergaard, M.; Jensen, J.P.; Jeppesen, E. Role of Sediment and Internal Loading of Phosphorus in Shallow Lakes. Hydrobiologia 2003, 506–509, 135–145. [Google Scholar] [CrossRef]
- Xu, R.; Lyu, T.; Zhang, M.; Cooper, M.; Pan, G. Molecular-Level Investigations of Effective Biogenic Phosphorus Adsorption by a Lanthanum/Aluminum-Hydroxide Composite. Sci. Total Environ. 2020, 725, 138424. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Lyu, T.; Wang, L.; Yuan, Y.; Zhang, M.; Cooper, M.; Mortimer, R.J.G.; Yang, Q.; Pan, G. Utilization of Coal Fly Ash Waste for Effective Recapture of Phosphorus from Waters. Chemosphere 2022, 287, 132431. [Google Scholar] [CrossRef] [PubMed]
- Haghseresht, F.; Wang, S.; Do, D.D. A Novel Lanthanum-Modified Bentonite, Phoslock, for Phosphate Removal from Wastewaters. Appl. Clay Sci. 2009, 46, 369–375. [Google Scholar] [CrossRef]
- Yin, H.; Kong, M.; Han, M.; Fan, C. Influence of Sediment Resuspension on the Efficacy of Geoengineering Materials in the Control of Internal Phosphorous Loading from Shallow Eutrophic Lakes. Environ. Pollut. 2016, 219, 568–579. [Google Scholar] [CrossRef]
- Hansen, A.M.; Ruiz-Castro, A.A.; Diáz-Valencia, S.E.; Moreno-Ayala, V.G.; Diáz-Aldama, E.; Sandoval, D.A.; Santana-Vega, Z. Dimensionamiento Hidrogeoquímico Para La Rehabilitación de Cuerpos de Agua. Acta INAGEQ 2020, 26, 9–29. (In Spanish) [Google Scholar]
- Gächter, R.; Müller, B. Why the Phosphorus Retention of Lakes Does Not Necessarily Depend on the Oxygen Supply to Their Sediment Surface. Limnol. Oceanogr. 2003, 48, 929–933. [Google Scholar] [CrossRef]
- Hansen, A.M.; Márquez-Pacheco, H. Procedimiento Para Evaluar Cargas Internas de Nutrientes En Cuerpos de Agua. Rev. Mex. Cienc. Geológicas 2012, 29, 265–275. [Google Scholar]
- Walthert, L.; Graf, U.; Kammer, A.; Luster, J.; Pezzotta, D.; Zimmermann, S.; Hagedorn, F. Determination of Organic and Inorganic Carbon, δ 13 C, and Nitrogen in Soils Containing Carbonates after Acid Fumigation with HCl. J. Plant Nutr. Soil Sci. 2010, 173, 207–216. [Google Scholar] [CrossRef]
- ASTM (American Society for Testing and Materials) Standard Test Methods for Determining the Water (Moisture) Content, Ash Content, and Organic Material of Peat and Other Organic Soils. ASTM D 2974-07a Standard Test. Available online: https://www.astm.org/d2974-20e01.html (accessed on 11 March 2022).
- Merck Test Alkalinity 111109. Available online: https://www.merckmillipore.com/MX/es/product/Alkalinity-Test,MDA_CHEM-111109#documentation (accessed on 11 September 2021).
- EPA (Environmental Protection Agency). Method 365.3: Phosphorous, All Forms (Colorimetric, Ascorbic Acid, Two Reagent). Available online: https://www.epa.gov/sites/default/files/2015-08/documents/method_365-3_1978.pdf (accessed on 15 September 2021).
- ISO (International Organization for Standardization). ISO 11905-1: Water Quality—Determination of Nitrogen—Part 1: Method Using Oxidative Digestion with Peroxodisulfate. Available online: https://www.iso.org/cms/render/live/en/sites/isoorg/contents/data/standard/00/21/2155.html (accessed on 27 September 2021).
- DIN (Deutsches Institut für Normung). DIN 38405 German Standard Methods for Examination of Water, Waste Water and Sludge—Anions (Group D)—Part 9: Spectrometric Determination of Nitrate (D 9). Available online: https://www.din.de/en/getting-involved/standards-committees/textilnorm/publications/wdc-beuth:din21:144185708 (accessed on 27 September 2021).
- DIN (Deutsches Institut für Normung). DIN 38406-5 German Standard Methods for the Examination of Water, Waste Water and Sludge; Cations (Group E); Determination of Ammonia-Nitrogen (E 5). Available online: https://www.din.de/en/getting-involved/standards-committees/textilnorm/publications/wdc-beuth:din21:1061756 (accessed on 27 September 2021).
- EPA (Environmental Protection Agency). Method—375.4. Methods and Guidance for Analysis of Water: Sulfate (Turbidimetric). Available online: https://www.nemi.gov/methods/method_summary/5316/ (accessed on 15 September 2021).
- EPA (Environmental Protection Agency). Method—376.2. Methods and Guidance for Analysis of Water: Sulfide (Colorimetric, Methylene Blue). Available online: https://www.nemi.gov/methods/method_summary/5318/ (accessed on 15 September 2021).
- APHA (American Public Health Association). 3500-Fe Iron. In Standard Methods for the Examination of Water and Wastewater; American Public Health Association: Washington, DC, USA, 2018. [Google Scholar]
- Merck Test Manganese-114770. Available online: https://www.merckmillipore.com/MX/es/product/Manganese-Test,MDA_CHEM-114770 (accessed on 11 September 2021).
- Merck, Calcium Test 14815. Available online: https://www.merckmillipore.com/MX/es/product/SQ-Calcium-Test,MDA_CHEM-114815 (accessed on 29 April 2022).
- Merck, Magnesium Cell Test 100815. Available online: https://www.merckmillipore.com/MX/es/product/Magnesium-Cell-Test,MDA_CHEM-100815 (accessed on 28 April 2022).
- Merck Sodium Cell Test 100885. Available online: https://www.merckmillipore.com/MX/es/product/Sodium-Cell-Test-in-nutrient-solutions-for-fertilization,MDA_CHEM-100885 (accessed on 28 April 2022).
- Merck, Potassium Cell Test. Available online: https://www.merckmillipore.com/MX/es/product/Potassium-Cell-Test,MDA_CHEM-114562 (accessed on 29 April 2021).
- Merck, Chloride Test 14897. Available online: https://www.merckmillipore.com/MX/es/product/Chloride-Test,MDA_CHEM-114562 (accessed on 28 April 2022).
- Di Rienzo, J.; Casanoves, F.; Balzarini, M.; González, L. Infostat—Software Estadístico; Universidad Nacional de Córdoba: Córdoba, Argentina, 2020. [Google Scholar]
- Hansen, A.M.; Hernández-Martínez, C.; Falcón-Rojas, A. Evaluation of Eutrophication Control Through Hypolimnetic Oxygenation. Procedia Earth Planet. Sci. 2017, 17, 598–601. [Google Scholar] [CrossRef]
- Hansen, A.M.; Márquez-Pacheco, H. Internal Phosphorus Load in a Mexican Reservoir: Forecast and Validation: Internal Phosphorus Load in a Mexican Reservoir. Environ. Toxicol. Chem. 2015, 34, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Phoslock Europe GmbH about Phoslock. Available online: https://www.phoslock.eu/es/sobre-phoslock (accessed on 21 May 2021).
- SRI Instrument. Method 25 Methane/Nonmethane GC. Available online: https://www.srigc.com/home/product_detail/method-25-methanenonmethane-gc (accessed on 12 August 2021).
- Haynes, W.M. (Ed.) CRC Handbook of Chemistry and Physics, 95th ed.; CRC Press: Boca Raton, FL, USA, 2014; ISBN 978-0-429-17019-5. [Google Scholar]
- Grundl, T.J.; Haderlein, S.; Nurmi, J.T.; Tratnyek, P.G. Introduction to Aquatic Redox Chemistry; ACS Symposium Series; Tratnyek, P.G., Grundl, T.J., Haderlein, S.B., Eds.; American Chemical Society: Washington, DC, USA, 2011; Volume 1071, pp. 1–14. ISBN 978-0-8412-2652-4. [Google Scholar]
- Burgin, A.J.; Yang, W.H.; Hamilton, S.K.; Silver, W.L. Beyond Carbon and Nitrogen: How the Microbial Energy Economy Couples Elemental Cycles in Diverse Ecosystems. Front. Ecol. Environ. 2011, 9, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Carnero-Bravo, V.; Merino-Ibarra, M.; Ruiz-Fernández, A.C.; Sanchez-Cabeza, J.A.; Ghaleb, B. Sedimentary Record of Water Column Trophic Conditions and Sediment Carbon Fluxes in a Tropical Water Reservoir (Valle de Bravo, Mexico). Environ. Sci. Pollut. Res. 2015, 22, 4680–4694. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.D. Diffuse Flux of Greenhouse Gases—Methane and Carbon Dioxide—At the Sediment-Water Interface of Some Lakes and Reservoirs of the World. In Greenhouse Gas Emissions—Fluxes and Processes; Tremblay, A., Varfalvy, L., Roehm, C., Garneau, M., Eds.; Environmental Science and Engineering; Springer: Berlin/Heidelberg, Germany, 2005; pp. 129–153. ISBN 978-3-540-23455-5. [Google Scholar]
- Bastviken, D. Methane. In Encyclopedia of Inland Waters; Elsevier: Amsterdam, The Netherlands, 2009; pp. 783–805. ISBN 978-0-12-370626-3. [Google Scholar]
- Stoeva, M.K.; Coates, J.D. Specific Inhibitors of Respiratory Sulfate Reduction: Towards a Mechanistic Understanding. Microbiology 2019, 165, 254–269. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Processes | Mineralization Reactions |
|---|---|
| O(Ø)/(-II) | (CH2O)106(NH3)16(PO43−) + 138O2 → 106CO2 + 16NO3− + PO43− + 122H2O |
| Mn(IV)/(II) | (CH2O)106(NH3)16(PO43−) + 236MnO2 + 472H+ → 236Mn2+ + 106CO2 + 8N2 + PO43− + 366H2O |
| N(V)/(Ø) | (CH2O)106(NH3)16(PO43−) + 94.4NO3− + 94.4H+ → 106CO2 + 55.2N2 + PO43− + 177.2H2O |
| Fe(III)/(II) | (CH2O)106(NH3)16(PO43−) + 424FeOOH + 848H+ → 424Fe2+ + 106CO2 + 16NH3 + PO43− + 742H2O |
| S(VI)/(-II) | (CH2O)106(NH3)16(PO43−) + 53SO42− → 106CO2 + 16NH3 + 53S2− + PO43− + 106H2O |
| C(Ø)/(±IV) | (CH2O)106(NH3)16(PO43−) → 53CO2 + 53CH4 + 16NH3 + PO43− |
| Parameter | Method | Reference |
|---|---|---|
| Total Organic Carbon (TOC) | ASTM D2974 | [47] |
| Alkalinity | MQuant 11109 | [48] |
| Phosphate (PO43−) | EPA 365.3 | [49] |
| Total Nitrogen (NT) | ISO 11905-1 | [50] |
| Nitrate (NO3−) | DIN 38405 | [51] |
| Ammonium (NH4+) | DIN 38406-5 | [52] |
| Sulfate (SO42−) | EPA 375.4 | [53] |
| Sulfide (S2−) | EPA 376.2 | [54] |
| Total iron (FeT), Fe (II and III) | APHA 3500-Fe | [55] |
| Manganese (II) | MQuant 14770 | [56] |
| Calcium | MQuant 14815 | [57] |
| Magnesium | MQuant 100815 | [58] |
| Sodium | MQuant 114562 | [59] |
| Potassium | MQuant 100885 | [60] |
| Chloride | MQuant 14897 | [61] |
| Reactor | Method | Gas |
|---|---|---|
| CONTROL | No eutrophication control treatment | 20 mL/20 min N2 (Infra 99.9%) as carrier gas |
| HOS | Intermittent applications of O2 (g) | 20 mL/20 min O2 (Infra 99.7%) for oxygenation and as carrier gas |
| PHOS | Intermittent applications of Phoslock | 20 mL/20 min N2 (Infra 99.9%) as carrier gas |
| HOS + PHOS | Intermittent applications of O2 (g) and Phoslock | 20 mL/20 min O2 (Infra 99.7%) for oxygenation and as carrier gas |
| Parameter | Initial | CONTROL | HOS | HOS + PHOS | PHOS |
|---|---|---|---|---|---|
| Emitted GHCG | 0 | 1262 | 1040 | 890 | 782 |
| GHCGaq | <1 | <1 | <1 | <1 | <1 |
| Alkalinity | 37 | 238 | 245 | 234 | 245 |
| TOCaq | 10 | 155 | 183 | 196 | 156 |
| TOCsed | 11,302 | 9591 | 9826 | 9912 | 10,067 |
| TICsed | <1 | <1 | <1 | <1 | <1 |
| Total | 11,349 | 11,246 | 11,294 | 11,232 | 11,250 |
| Reactor | Gas-and Nutrient Application Events | |||||
|---|---|---|---|---|---|---|
| 1–2 (0–60 d) | 2–3 (60–74 d) | 3–4 (74–81 d) | ||||
| Eh | pH | Eh | pH | Eh | pH | |
| CONTROL | 282 ± 203 | 4.7 ± 0.8 | −260 ± 175 | 6.0 ± 0.2 | −477 ± 34 | 6.2 ± 0.2 |
| HOS | 131 ± 240 | 4.7 ± 0.6 | −348 ± 73 | 6.1 ± 0.1 | −450 ± 4 | 6.3 ± 0.1 |
| HOS + PHOS | 24 ± 339 | 4.8 ± 1.1 | −42 ± 171 | 6.0 ± 0.2 | −253 ± 390 | 6.2 ± 0.1 |
| PHOS | 216 ± 40 | 5.5 ± 0.3 | −339 ± 100 | 6.3 ± 0.2 | −466 ± 14 | 6.3 ± 0.2 |
| Average | 161 ± 242 | 4.9 ± 0.8 | −212 ± 232 | 6.1 ± 0.2 | −409 ± 171 | 6.2 ± 0.1 |
| Reactor | Time | TOC | Alk | PO43− | NO3− | NH4+ | N2 | SO42− | S2− | Fe(T) | Fe(III) | Fe(II) | Mn(II) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (d) | mmol m−2 | ||||||||||||
| CONTROL | 0 | 10 | 108 | 0.07 | 15.4 | 14.1 | 0 | 123 | <0.06 | 7.049 | 0.065 | 6.984 | 1.78 |
| 60 | 75 | 152 | 0.23 | 0.51 | 11.2 | 73.2 | 637 | 0.06 | 1.072 | 0.520 | 0.552 | 0.71 | |
| 74 | 152 | 175 | 0.24 | 0.25 | 24.6 | 26.1 | 279 | 0.07 | 0.585 | 0.032 | 0.552 | 0.60 | |
| 81 | 155 | 234 | 1.18 | 0.25 | 19.5 | 6.3 | 0 | 0.65 | 4.467 | <0.001 | 4.467 | 0.45 | |
| HOS | 0 | 10 | 78 | 0.08 | 30.6 | 17.9 | 0 | 58 | <0.06 | 0.474 | 0.058 | 0.416 | 1.68 |
| 60 | 114 | 138 | 1.67 | 0.51 | 13.9 | 81.3 | 491 | 0.09 | 11.50 | <0.001 | 11.50 | 8.58 | |
| 74 | 179 | 204 | 0.11 | 0.25 | 21.1 | 13.9 | 299 | 0.11 | 0.780 | 0.065 | 0.715 | 0.66 | |
| 81 | 183 | 245 | 5.07 | 0.25 | 12.3 | 0 | 16 | 0.11 | 24.85 | <0.001 | 24.85 | 2.58 | |
| HOS + PHOS | 0 | 10 | 45 | 0.09 | 37.3 | 20.2 | 0 | 45 | <0.06 | 1.754 | 0.578 | 1.176 | 1.24 |
| 60 | 29 | 178 | 0.04 | 0.51 | 10.7 | 26.2 | 448 | 0.04 | 0.520 | 0.065 | 0.455 | 0.77 | |
| 74 | 165 | 197 | 0.08 | 2.55 | 18.1 | 32.4 | 309 | 0.20 | 31.25 | <0.001 | 31.25 | 2.82 | |
| 81 | 196 | 238 | 0.11 | 0.25 | 18.6 | 5.1 | 0.0 | 0.07 | 26.31 | <0.001 | 26.31 | 2.80 | |
| PHOS | 0 | 10 | 37 | 0.07 | 34.5 | 21.0 | 0 | 55 | <0.06 | 0.510 | 0.094 | 0.416 | 1.10 |
| 60 | 32 | 138 | 0.02 | 0.51 | 16.0 | 8.2 | 318 | 0.06 | 0.520 | 0.097 | 0.422 | 0.77 | |
| 74 | 121 | 230 | 0.02 | 6.37 | 24.1 | 10.2 | 263 | 0.01 | 6.205 | 0.162 | 6.042 | 1.09 | |
| 81 | 156 | 245 | 0.23 | 0.25 | 25.6 | 0 | 0.0 | 0.49 | 8.527 | <0.001 | 8.527 | 1.23 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval-Chacón, D.A.; Hansen, A.M. Effect of Eutrophication Control Methods on the Generation of Greenhouse Carbon Gases in Sediment. Water 2022, 14, 1705. https://doi.org/10.3390/w14111705
Sandoval-Chacón DA, Hansen AM. Effect of Eutrophication Control Methods on the Generation of Greenhouse Carbon Gases in Sediment. Water. 2022; 14(11):1705. https://doi.org/10.3390/w14111705
Chicago/Turabian StyleSandoval-Chacón, DAngelo A., and Anne M. Hansen. 2022. "Effect of Eutrophication Control Methods on the Generation of Greenhouse Carbon Gases in Sediment" Water 14, no. 11: 1705. https://doi.org/10.3390/w14111705
APA StyleSandoval-Chacón, D. A., & Hansen, A. M. (2022). Effect of Eutrophication Control Methods on the Generation of Greenhouse Carbon Gases in Sediment. Water, 14(11), 1705. https://doi.org/10.3390/w14111705