Aqueous Mercury Removal with Carbonaceous and Iron Sulfide Sorbents and Their Applicability as Thin-Layer Caps in Mercury-Contaminated Estuary Sediment



Abstract
1. Introduction
2. Materials and Methods
2.1. Physicochemical Properties of Sorbents and Sediment
2.2. Aqueous Batch Sorption Experiment
2.2.1. Preparation of Hg Stock Solution
2.2.2. Sample Preparation
- (1)
- The Hg stock solution of 50 mL was injected into the glass bottles.
- (2)
- The intended dosage of sorbents was added into the glass bottles and sealed with a rubber plug and aluminum cap.
- (3)
- The bottles were collected, including the samples of triplicate plus the blank.
2.2.3. Adsorption Process
2.2.4. Preservation of Sample and Analysis
- (1)
- Each sample was preserved with 0.5% BrCl2 and estimated by cold vapor atomic fluorescence spectroscopy (CVAFS; Brooks Rand Automated Total Mercury System, Seattle, WA, USA).
- (2)
- The aqueous Hg removal efficiency by sorbents was calculated by the following Equation (1):where R (%) is the Hg removal efficiency of sorbents, C0 (ng/L) is the initial Hg concentrations detected in blank solution, and Ct (ng/L) is the concentration of remaining Hg at any time.
2.3. Laboratory Microcosm Experiments
2.4. Statistical Analysis
3. Results and Discussion
3.1. Physicochemical Properties of Sorbents
3.2. Aqueous Batch Adsorption Experiment
3.2.1. The Effects of the Sorbent Dosage
3.2.2. Effect of Initial Concentration of Hg2+
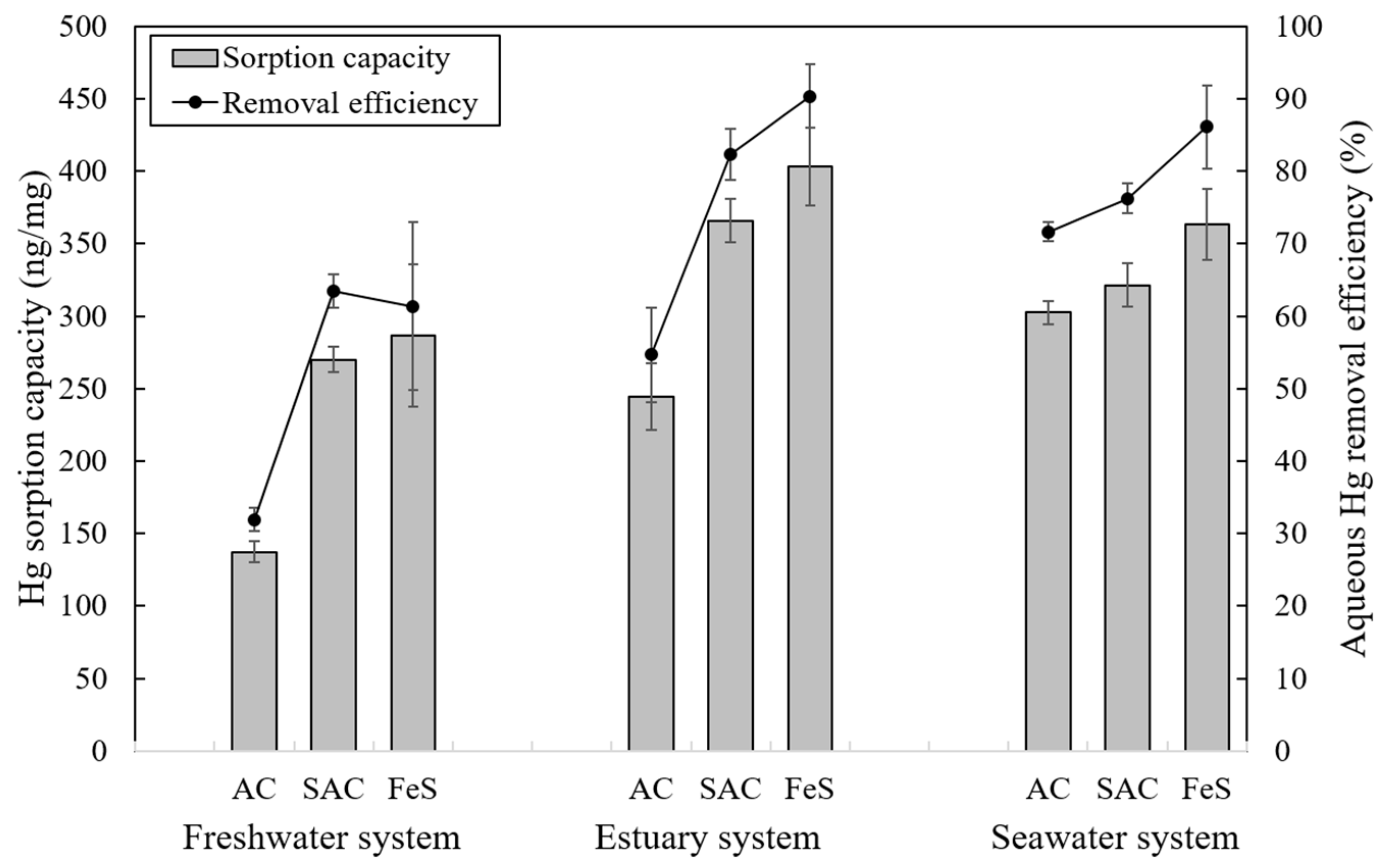
3.2.3. Effect of Salinity
3.2.4. Effect of Dissolved Organic Matter on Hg Sorption at Various Salinity Levels
3.3. Laboratory Microcosm Experiments
3.3.1. Physicochemical Properties of Sediments Used in Microcosm Experiments
3.3.2. Environmental Factor Variation during Microcosms Operation
3.3.3. Sequestration of Aqueous THg and MeHg by Thin Layer Capping
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nriagu, J.O. Biogeochemistry of Mercury in the Environment; Elsevier/North-Holland Biomedical Press: Amsterdam, The Netherlands, 1979. [Google Scholar]
- Compeau, G.C.; Bartha, R. Sulfate-reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Appl. Environ. Microbiol. 1985, 50, 498–502. [Google Scholar] [CrossRef]
- Gilmour, C.C.; Henry, E.A.; Mitchell, R. Sulfate stimulation of mercury methylation in freshwater sediments. Environ. Sci. Technol. 1992, 26, 2281–2287. [Google Scholar] [CrossRef]
- Gilmour, C.C.; Podar, M.; Bullock, A.L.; Graham, A.M.; Brown, S.D.; Somenahally, A.C.; Johs, A.; Hurt, R.A.; Bailey, K.L.; Elias, D.A. Mercury methylation by novel microorganisms from new environments. Environ. Sci. Technol. 2013, 47, 11810–11820. [Google Scholar] [CrossRef] [PubMed]
- Podar, M.; Gilmour, C.C.; Brandt, C.C.; Soren, A.; Brown, S.D.; Crable, B.R.; Palumbo, A.V.; Somenahally, A.C.; Elias, D.A. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci. Adv. 2015, 1, e1500675. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, W.F.; Lamborg, C.H.; Hammerschmidt, C.R. Marine biogeochemical cycling of mercury. Chem. Rev. 2007, 107, 641–662. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.D.; Cohen, Y.; Winer, A.M. Environmental distribution and transformation of mercury compounds. Crit. Rev. Environ. Sci. Technol. 1996, 26, 1–43. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, M.Y.; Zeng, G.M.; Yu, Z.G.; Cui, F.; Yang, Z.Z.; Shen, L.Q. Active capping technology: A new environmental remediation of contaminated sediment. Environ. Sci. Pollut. Res. 2016, 23, 4370–4386. [Google Scholar] [CrossRef] [PubMed]
- McDonough, K.M.; Murphy, P.; Olsta, J.; Zhu, Y.; Reible, D.; Lowry, G.V. Development and placement of a sorbent-amended thin layer sediment cap in the Anacostia River. Soil Sediment Contam. 2007, 16, 313–322. [Google Scholar] [CrossRef]
- Gilmour, C.; Bell, T.; Soren, A.; Riedel, G.; Riedel, G.; Kopec, D.; Bodaly, D.; Ghosh, U. Activated carbon thin-layer placement as an in situ mercury remediation tool in a Penobscot River salt marsh. Sci. Total Environ. 2018, 621, 839–848. [Google Scholar] [CrossRef]
- Ghosh, U.; Luthy, R.G.; Cornelissen, G.; Werner, D.; Menzie, C.A. In-situ sorbent amendments: A new direction in contaminated sediment management. Environ. Sci. Technol. 2011, 45, 1163–1168. [Google Scholar] [CrossRef]
- Gomez-Eyles, J.L.; Yu, P.Q.; Beckingham, B.; Riedel, G.; Gilmour, C.C.; Ghosh, U. Evaluation of biochars and activated carbons for in situ remediation of sediments impacted with organics, mercury, and methylmercury. Environ. Sci. Technol. 2013, 47, 13721–13729. [Google Scholar] [CrossRef] [PubMed]
- Ie, I.R.; Hung, C.H.; Jen, Y.S.; Yuan, C.S.; Chen, W.H. Adsorption of vapor-phase elemental mercury (Hg0) and mercury chloride (HgCl2) with innovative composite activated carbons impregnated with Na2S and S0 in different sequences. Chem. Eng. J. 2013, 229, 469–476. [Google Scholar] [CrossRef]
- Puri, B.R.; Hazra, R.S. Carbon-sulphur surface complexes on charcoal. Carbon 1971, 9, 123–134. [Google Scholar] [CrossRef]
- Li, Z.; Wu, L.; Liu, H.; Lan, H.; Qu, J. Improvement of aqueous mercury adsorption on activated coke by thiol-functionalization. Chem. Eng. J. 2013, 228, 925–934. [Google Scholar] [CrossRef]
- Macıas-Garcıa, A.; Gomez-Serrano, V.; Alexandre-Franco, M.F.; Valenzuela-Calahorro, C. Adsorption of cadmium by sulphur dioxide treated activated carbon. J. Hazard. Mater. 2003, 103, 141–152. [Google Scholar] [CrossRef]
- Ting, Y.; Chen, C.; Ch’ng, B.L.; Wang, Y.L.; Hsi, H.C. Using raw and sulfur-impregnated activated carbon as active cap for leaching inhibition of mercury and methylmercury from contaminated sediment. J. Hazard. Mater. 2018, 354, 116–124. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, Y.; Xiong, Z.; Kaback, D.; Zhao, D. Immobilization of mercury in field soil and sediment using carboxymethyl cellulose stabilized iron sulfide nanoparticles. Nanotechnology 2012, 23, 294007. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, Y.; Xiong, Z.; Zhao, D. Immobilization of mercury by carboxymethyl cellulose stabilized iron sulfide nanoparticles: Reaction mechanisms and effects of stabilizer and water chemistry. Environ. Sci. Technol. 2014, 48, 3986–3994. [Google Scholar] [CrossRef]
- Han, D.S.; Orillano, M.; Khodary, A.; Duan, Y.; Batchelor, B.; Abdel-Wahab, A. Reactive iron sulfide (FeS)-supported ultrafiltration for removal of mercury (Hg (II)) from water. Water Res. 2014, 53, 310–321. [Google Scholar] [CrossRef]
- Liu, J.; Valsaraj, K.T.; Devai, I.; DeLaune, R.E. Immobilization of aqueous Hg (II) by mackinawite (FeS). J. Hazard. Mater. 2008, 157, 432–440. [Google Scholar] [CrossRef]
- Xiong, Z.; He, F.; Zhao, D.; Barnett, M.O. Immobilization of mercury in sediment using stabilized iron sulfide nanoparticles. Water Res. 2009, 43, 5171–5179. [Google Scholar] [CrossRef] [PubMed]
- Paquette, K.; Helz, G. Solubility of cinnabar (red HgS) and implications for mercury speciation in sulfidic waters. Water Air Soil Poll. 1995, 80, 1053–1056. [Google Scholar] [CrossRef]
- Benoit, J.M.; Gilmour, C.C.; Mason, R.P.; Heyes, A. Sulfide controls on mercury speciation and bioavailability to methylating bacteria in sediment pore waters. Environ. Sci. Technol. 1999, 33, 951–957. [Google Scholar] [CrossRef]
- Ravichandran, M.; Aiken, G.R.; Ryan, J.N.; Reddy, M.M. Inhibition of precipitation and aggregation of metacinnabar (mercuric sulfide) by dissolved organic matter isolated from the Florida Everglades. Environ. Sci. Technol. 1999, 33, 1418–1423. [Google Scholar] [CrossRef]
- Morse, J.W.; Arakaki, T. Adsorption and coprecipitation of divalent metals with mackinawite (FeS). Geochim. Cosmochim. Acta 1993, 57, 3635–3640. [Google Scholar] [CrossRef]
- Wolthers, M.; Van der Gaast, S.J.; Rickard, D. The structure of disordered mackinawite. Am. Mineral. 2003, 88, 2007–2015. [Google Scholar] [CrossRef]
- Hsi, H.C.; Rood, M.J.; Rostam-Abadi, M.; Chen, S.; Chang, R. Effects of sulfur impregnation temperature on the properties and mercury adsorption capacities of activated carbon fibers (ACFs). Environ. Sci. Technol. 2001, 35, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.J.; Chiou, H.J.; Chen, Y.H.; Lin, K.S.; Rood, M.J.; Hsi, H.C. Mercury adsorption and re-emission inhibition from actual WFGD wastewater using sulfur-containing activated carbon. Environ. Res. 2019, 168, 319–328. [Google Scholar] [CrossRef]
- ASTM D6556–10. Standard Test Method for Carbon Black—Total and External Surface Area by Nitrogen Adsorption; ASTM International: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Lippens, B.C.; de Boer, J.H. Studies on pore systems in catalysts: V. the t method. J. Catal. 1965, 4, 319–323. [Google Scholar] [CrossRef]
- Gee, G.W.; Bauder, J.W. Particle-size analysis. In Methods of Soil Analysis, Part 1. Physical and Mineralogical Methods, 2nd ed.; Klute, A., Ed.; Agronomy Monograph No. 9; American Society of Agronomy/Soil Science Society of America: Madison, WI, USA, 1986; pp. 383–411. [Google Scholar]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon, and organic matter. In Methods of Soil Analysis, Part 2. Chemical and Microbiological Properties, 2nd ed.; Page, A.L., Ed.; Agronomy Series No. 9; American Society of Agronomy/Soil Science Society of America: Madison, WI, USA, 1982; pp. 539–579. [Google Scholar]
- Lewis, P.A.; Klemm, D.J.; Lazorchak, J.M.; Norberg-King, T.J.; Peltier, W.H.; Heber, M.A. Short-Term Methods for Estimating the Chronic Toxicity of Effluents and Receiving Waters to Freshwater Organisms; US Environmental Protection Agency, Environmental Monitoring Systems Laboratory: Cincinnati, OH, USA, 1994.
- Kester, D.R.; Duedall, I.W.; Connors, D.N.; Pytkowicz, R.M. Preparation of artificial seawater 1. Limnol. Oceanogr. 1967, 12, 176–179. [Google Scholar] [CrossRef]
- Wang, Y.L.; Fang, M.D.; Chien, L.C.; Lin, C.C.; Hsi, H.C. Distribution of mercury and methylmercury in surface water and surface sediment of river, irrigation canal, reservoir and wetland in Taiwan. Environ. Sci. Pollut. Res. 2019, 26, 17762–17773. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, F.; Younesi, H.; Ghoreyshi, A.A.; Bahramifar, N.; Heidari, A. Thiol-incorporated activated carbon derived from fir wood sawdust as an efficient adsorbent for the removal of mercury ion: Batch and fixed-bed column studies. Process Saf. Environ. Prot. 2016, 100, 22–35. [Google Scholar] [CrossRef]
- de Diego, A.; Tseng, C.M.; Dimov, N.; Amouroux, D.; Donard, O.F. Adsorption of aqueous inorganic mercury and methylmercury on suspended kaolin: Influence of sodium chloride, fulvic acid and particle content. Appl. Organomet. Chem. 2001, 15, 490–498. [Google Scholar] [CrossRef]
- Ranganathan, K. Adsorption of Hg (II) ions from aqueous chloride solutions using powdered activated carbons. Carbon 2003, 41, 1087–1092. [Google Scholar] [CrossRef]
- Thiem, L.; Badorek, D.; O’Connor, J.T. Removal of mercury from drinking water using activated carbon. J. Am. Water Works Assoc. 1976, 68, 447–451. [Google Scholar] [CrossRef]
- Moreno, F.N.; Anderson, C.W.; Stewart, R.B.; Robinson, B.H.; Ghomshei, M.; Meech, J.A. Induced plant uptake and transport of mercury in the presence of sulphur-containing ligands and humic acid. New Phytol. 2005, 166, 445–454. [Google Scholar] [CrossRef]
- Muller, K.A.; Brandt, C.C.; Mathews, T.J.; Brooks, S.C. Methylmercury sorption onto engineered materials. J. Environ. Manag. 2019, 245, 481–488. [Google Scholar] [CrossRef]
- Schwartz, G.E.; Sanders, J.P.; McBurney, A.M.; Brown, S.S.; Ghosh, U.; Gilmour, C.C. Impact of dissolved organic matter on mercury and methylmercury sorption to activated carbon in soils: Implications for remediation. Environ. Sci. Process. Impacts 2019, 21, 485–496. [Google Scholar] [CrossRef]
- Johs, A.; Eller, V.A.; Mehlhorn, T.L.; Brooks, S.C.; Harper, D.P.; Mayes, M.A.; Pierce, E.M.; Peterson, M.J. Dissolved organic matter reduces the effectiveness of sorbents for mercury removal. Sci. Total Environ. 2019, 690, 410–416. [Google Scholar] [CrossRef]
- Skyllberg, U.; Bloom, P.R.; Qian, J.; Lin, C.M.; Bleam, W.F. Complexation of mercury (II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups. Environ. Sci. Technol. 2006, 40, 4174–4180. [Google Scholar] [CrossRef]
- Skyllberg, U.; Drott, A. Competition between disordered iron sulfide and natural organic matter associated thiols for mercury (II): An EXAFS study. Environ. Sci. Technol. 2010, 44, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Pandey, S.D.; Misra, V. Stability constants of metal–humic acid complexes and its role in environmental detoxification. Ecotoxicol. Environ. Saf. 2000, 47, 195–200. [Google Scholar] [CrossRef]
- Ting, Y.; Ch’ng, B.L.; Chen, C.; Ou, M.Y.; Cheng, Y.H.; Hsu, C.J.; Hsi, H.C. A simulation study of mercury immobilization in estuary sediment microcosm by activated carbon/clay-based thin-layer capping under artificial flow and turbation. Sci. Total Environ. 2020, 708, 135068. [Google Scholar] [CrossRef] [PubMed]
- Compeau, G.C.; Bartha, R. Effect of salinity on mercury-methylating activity of sulfate-reducing bacteria in estuarine sediments. Appl. Environ. Microbiol. 1987, 53, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.M.; Tanton, T.W.; Abdrashitova, S.A. Mercury in the aquatic environment: A review of factors affecting methylation. Crit. Rev. Environ. Sci. Technol. 2001, 31, 241–293. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SBET (m2/g) * | SMicro (m2/g) * | VTotal (cm3/g) * | Vmicro (cm3/g) * | ||
| AC | 1024.1 | 634.1 | 0.540 | 0.284 | |
| SAC | 903.3 | 528.9 | 0.502 | 0.267 | |
| FeS | 2.811 | 0.356 | 0.04 | - | |
| C (%) | H (%) | O (%) | N (%) | S (%) | |
| AC | 78.3 | 1.61 | 7.72 | 0.791 | 0.672 |
| SAC | 74.9 | 1.80 | 13.8 | 0.36 | 5.75 |
| FeS | - | - | - | - | 36.4 |
| Parameters | Value |
|---|---|
| Water content (wt%) | 2.33 ± 0.01 |
| pH | 7.5 ± 0.03 |
| Organic carbon (wt%) | 0.80 ± 0.65 |
| Cation exchange capacity (cmol(+)/kg) | 3.3 ± 0.2 |
| Sand (wt%) | 71.4 ± 2.9 |
| Silt (wt%) | 14.3 ± 0.0 |
| Clay (wt%) | 14.3 ± 2.9 |
| THg (mg/kg) | 76.0 ± 2.59 |
| MeHg (μg/kg) | 1.17 ± 0.31 |
| Sediment 1 | Sorbents | Overlying Water | Recovery 2 | |||
|---|---|---|---|---|---|---|
| mg/kg | mg | mg/kg | mg | mg | % | |
| F-AC | 65.35 ± 0.65 | 19.61 | 4.84 ± 0.81 | 0.04 | 0.002 | 86.19 |
| F-SAC | 68.87 ± 0.46 | 20.66 | 6.86 ± 3.52 | 0.06 | 0.02 | 90.89 |
| F-FeS | 50.38 ± 1.03 | 15.11 | 37.66 ± 8.34 | 0.34 | 0.02 | 67.78 |
| F-Control | 68.77 ± 2.03 | 20.63 | - | - | 0.02 | 90.50 |
| E-AC | 56.16 ± 2.87 | 16.85 | 3.88 ± 1.82 | 0.03 | 0.002 | 74.05 |
| E-SAC | 57.55 ± 0.39 | 17.27 | 2.89 ± 1.49 | 0.03 | 0.003 | 75.85 |
| E-FeS | 54.80 ± 2.25 | 16.44 | 43.83 ± 13.04 | 0.39 | 0.002 | 73.84 |
| E-Control | 69.20 ± 4.02 | 20.70 | - | - | 0.002 | 90.80 |
| Freshwater System | MeHg (μg/kg) | Estuary System | MeHg (μg/kg) |
|---|---|---|---|
| AC | 1.43 ± 0.75 | AC | 1.03 ± 0.52 |
| SAC | 5.42 ± 1.92 | SAC | 1.16 ± 0.07 |
| FeS | 2.61 ± 0.65 | FeS | 2.07 ± 0.52 |
| Control unit | 3.97 ± 1.68 | Control unit | 1.99 ± 1.15 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ch’ng, B.-L.; Hsu, C.-J.; Ting, Y.; Wang, Y.-L.; Chen, C.; Chang, T.-C.; Hsi, H.-C. Aqueous Mercury Removal with Carbonaceous and Iron Sulfide Sorbents and Their Applicability as Thin-Layer Caps in Mercury-Contaminated Estuary Sediment. Water 2020, 12, 1991. https://doi.org/10.3390/w12071991
Ch’ng B-L, Hsu C-J, Ting Y, Wang Y-L, Chen C, Chang T-C, Hsi H-C. Aqueous Mercury Removal with Carbonaceous and Iron Sulfide Sorbents and Their Applicability as Thin-Layer Caps in Mercury-Contaminated Estuary Sediment. Water. 2020; 12(7):1991. https://doi.org/10.3390/w12071991
Chicago/Turabian StyleCh’ng, Boon-Lek, Che-Jung Hsu, Yu Ting, Ying-Lin Wang, Chi Chen, Tien-Chin Chang, and Hsing-Cheng Hsi. 2020. "Aqueous Mercury Removal with Carbonaceous and Iron Sulfide Sorbents and Their Applicability as Thin-Layer Caps in Mercury-Contaminated Estuary Sediment" Water 12, no. 7: 1991. https://doi.org/10.3390/w12071991
APA StyleCh’ng, B.-L., Hsu, C.-J., Ting, Y., Wang, Y.-L., Chen, C., Chang, T.-C., & Hsi, H.-C. (2020). Aqueous Mercury Removal with Carbonaceous and Iron Sulfide Sorbents and Their Applicability as Thin-Layer Caps in Mercury-Contaminated Estuary Sediment. Water, 12(7), 1991. https://doi.org/10.3390/w12071991