
AKR1C1 as a Biomarker for Differentiating the Biological Effects of Combustible from Non-Combustible Tobacco Products

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Tobacco Product Preparations and Chemical Analysis
2.3. Cell Lines and Cultures
2.4. Treatments
2.5. Microarray Gene Expression Profiling
2.6. Gene Set Enrichment Analysis
2.7. Quantitative RT-PCR
3. Results
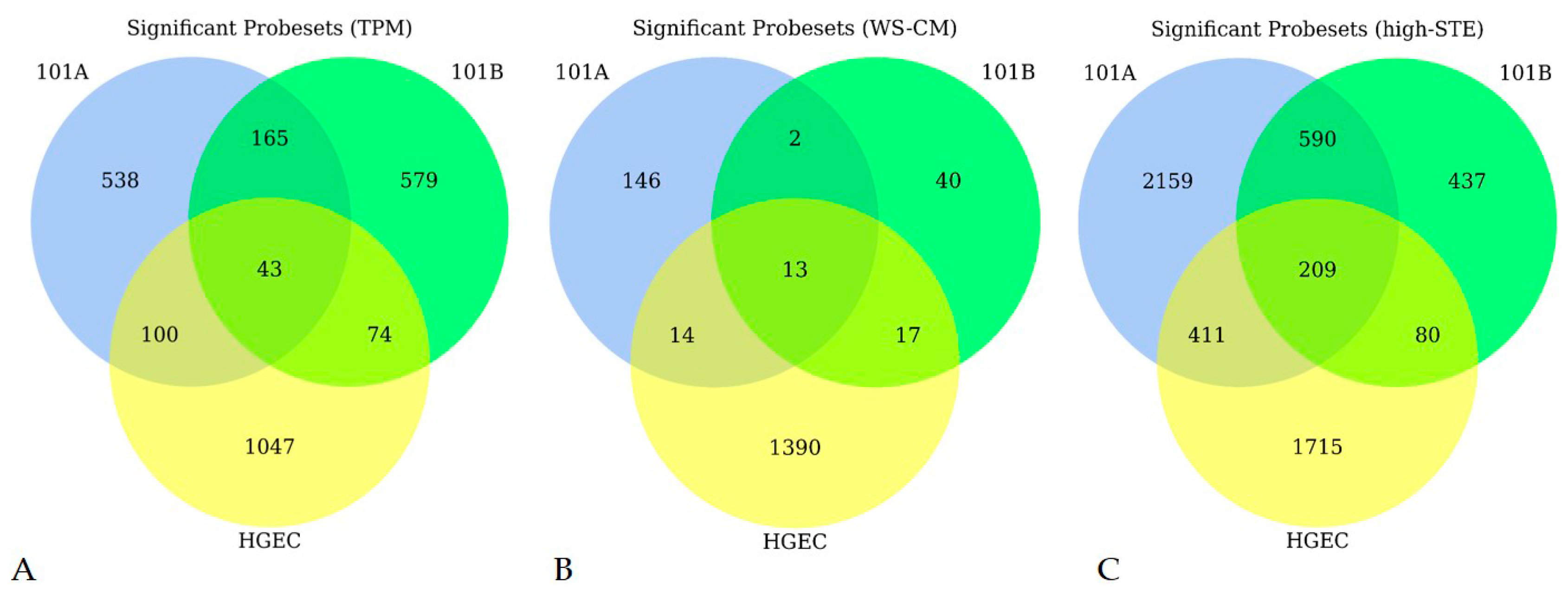
3.1. Microarray Expression Profiling
3.2. Gene Set Enrichment Analysis
3.3. AKR1C1 and AKR1C2 as Candidate Biomarkers for the Effects of Combustible Tobacco Products
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Jha, P.; Ramasundarahettige, C.; Landsman, V.; Rostron, B.; Thun, M.; Anderson, R.N.; McAfee, T.; Peto, R. 21st-century hazards of smoking and benefits of cessation in the United States. N. Engl. J. Med. 2013, 368, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.N.; Hamling, J. Systematic review of the relation between smokeless tobacco and cancer in Europe and North America. BMC Med. 2009, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Report, S.G. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2010.
- Henley, S.J.; Thun, M.J.; Connell, C.; Calle, E.E. Two large prospective studies of mortality among men who use snuff or chewing tobacco (United States). Cancer Causes Control 2005, 16, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.N. Epidemiological evidence relating snus to health--an updated review based on recent publications. Harm Reduct. J. 2013, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Piano, M.R.; Benowitz, N.L.; Fitzgerald, G.A.; Corbridge, S.; Heath, J.; Hahn, E.; Pechacek, T.F.; Howard, G. Impact of smokeless tobacco products on cardiovascular disease: Implications for policy, prevention, and treatment: A policy statement from the American Heart Association. Circulation 2010, 122, 1520–1544. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Hatsukami, D. The Strategic Dialogue on Tobacco Harm Reduction: A vision and blueprint for action in the US. Tob. Control 2009, 18, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J.; Phillips, L.D.; Balfour, D.; Curran, H.V.; Dockrell, M.; Foulds, J.; Fagerstrom, K.; Letlape, K.; Milton, A.; Polosa, R.; et al. Estimating the harms of nicotine-containing products using the MCDA approach. Eur. Addict. Res. 2014, 20, 218–225. [Google Scholar] [CrossRef] [PubMed]
- DeMarini, D.M. Genotoxicity of tobacco smoke and tobacco smoke condensate: A review. Mutat. Res. 2004, 567, 447–474. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H. Selective clonal expansion and microenvironmental permissiveness in tobacco carcinogenesis. Oncogene 2002, 21, 7392–7411. [Google Scholar] [CrossRef] [PubMed]
- Fields, W.R.; Leonard, R.M.; Odom, P.S.; Nordskog, B.K.; Ogden, M.W.; Doolittle, D.J. Gene expression in normal human bronchial epithelial (NHBE) cells following in vitro exposure to cigarette smoke condensate. Toxicol. Sci. 2005, 86, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Nordskog, B.K.; Blixt, A.D.; Morgan, W.T.; Fields, W.R.; Hellmann, G.M. Matrix-degrading and pro-inflammatory changes in human vascular endothelial cells exposed to cigarette smoke condensate. Cardiovasc. Toxicol. 2003, 3, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Semlali, A.; Witoled, C.; Alanazi, M.; Rouabhia, M. Whole cigarette smoke increased the expression of TLRs, HBDs, and proinflammory cytokines by human gingival epithelial cells through different signaling pathways. PLoS ONE 2012, 7, e52614. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Aggarwal, B.B. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-κB by suppressing activation of I–κB α kinase in human non-small cell lung carcinoma: Correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res. 2004, 64, 5004–5012. [Google Scholar] [PubMed]
- Arimilli, S.; Damratoski, B.E.; Bombick, B.; Borgerding, M.F.; Prasad, G.L. Evaluation of cytotoxicity of different tobacco product preparations. Regul. Toxicol. Pharmacol. 2012, 64, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Prasad, G.L.; Zacharias, W. Differential cell-specific cytotoxic responses of oral cavity cells to tobacco preparations. Toxicol. In Vitro 2013, 27, 282–291. [Google Scholar] [CrossRef] [PubMed]
- FDA. Harmful and Potentially Harmful Constituents in Tobacco Products and Tobacco Smoke; Established List; National Archives & Records Service; FDA: Silver Spring, MD, USA, 2012; Volume 77, pp. 20036–20037.
- Beranic, N.; Brozic, P.; Brus, B.; Sosic, I.; Gobec, S.; Lanisnik Rizner, T. Expression of human aldo-keto reductase 1C2 in cell lines of peritoneal endometriosis: Potential implications in metabolism of progesterone and dydrogesterone and inhibition by progestins. J. Steroid Biochem. Mol. Biol. 2012, 130, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Brozic, P.; Turk, S.; Rizner, T.L.; Gobec, S. Inhibitors of aldo-keto reductases AKR1C1-AKR1C4. Curr. Med. Chem. 2011, 18, 2554–2565. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Metabolism of chemical carcinogens. Carcinogenesis 2000, 21, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: A bio-historical perspective with updates. Carcinogenesis 2001, 22, 1903–1930. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Berlin, J.A.; Penning, T.M.; Field, J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem. Res. Toxicol. 2002, 15, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Aldo-Keto Reductase Regulation by the Nrf2 System: Implications for Stress Response, Chemotherapy Drug Resistance, and Carcinogenesis. Chem. Res. Toxicol. 2016, 30, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Prasad, G.L.; Zacharias, W. Combusted but not smokeless tobacco products cause DNA damage in oral cavity cells. Environ. Toxicol. Pharmacol. 2014, 37, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.O.; Gumus, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. (Phila) 2010, 3, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Gumus, Z.H.; Du, B.; Kacker, A.; Boyle, J.O.; Bocker, J.M.; Mukherjee, P.; Subbaramaiah, K.; Dannenberg, A.J.; Weinstein, H. Effects of tobacco smoke on gene expression and cellular pathways in a cellular model of oral leukoplakia. Cancer Prev. Res. (Phila) 2008, 1, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Severino, P.; Alvares, A.M.; Michaluart, P., Jr.; Okamoto, O.K.; Nunes, F.D.; Moreira-Filho, C.A.; Tajara, E.H. Head and Neck Genome Project GENCAPO Global gene expression profiling of oral cavity cancers suggests molecular heterogeneity within anatomic subsites. BMC Res. Notes 2008, 1, 113. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Schembri, F.; Zeskind, J.; Shah, V.; Gustafson, A.M.; Steiling, K.; Liu, G.; Dumas, Y.M.; Zhang, X.; Brody, J.S.; et al. Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genom. 2008, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Pappas, R.S.; Stanfill, S.B.; Watson, C.H.; Ashley, D.L. Analysis of toxic metals in commercial moist snuff and Alaskan iqmik. J. Anal. Toxicol. 2008, 32, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Rickert, W.S.; Joza, P.J.; Trivedi, A.H.; Momin, R.A.; Wagstaff, W.G.; Lauterbach, J.H. Chemical and toxicological characterization of commercial smokeless tobacco products available on the Canadian market. Regul. Toxicol. Pharmacol. 2009, 53, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔ Ct Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.; Yamauchi, N.; Moriguchi, H.; Hippo, Y.; Watanbe, A.; Shibahara, J.; Taniguichi, H.; Ishikawa, S.; Ito, H.; Yamamato, S.; et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers non-small cell lung carcinoma. Clin. Cancer Res. 2005, 11, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.; Ho, H.; Chow, K.; Lin, T.; Shih, C.; Wang, L.; Tsai, C. Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res. 2001, 61, 2727–2731. [Google Scholar] [PubMed]
- Nagaraj, N.S.; Beckers, S.; Mensah, J.K.; Waigel, S.; Vigneswaran, N.; Zacharias, W. Cigarette smoke condensate induces cytochromes P450 and aldo-keto reductases in oral cancer cells. Toxicol. Lett. 2006, 165, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lee, J.J.; Tang, H.; Fan, Y.H.; Xiao, L.; Ren, H.; Kurie, J.; Morice, R.C.; Hong, W.K.; Mao, L. Impact of smoking cessation on global gene expression in the bronchial epithelium of chronic smokers. Cancer Prev. Res. 2008, 1, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sebastiani, P.; Liu, G.; Schembri, F.; Dumas, Y.M.; Langer, E.M.; Alekseyev, Y.; O'Connor, G.T.; Brooks, D.R.; Lenburg, M.E.; et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol. Genom. 2010, 41, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Drury, J.E. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007, 464, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.W.; Reichardt, J.K. Modeling single nucleotide polymorphisms in the human AKR1C1 and AKR1C2 genes: Implications for functional and genotyping analyses. PLoS ONE 2010, 5, e15604. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Duan, L.; Lee, S.H.; Kloosterboer, H.J.; Blair, I.A.; Penning, T.M. Human cytosolic hydroxysteroid dehydrogenases of the aldo-ketoreductase superfamily catalyze reduction of conjugated steroids: Implications for phase I and phase II steroid hormone metabolism. J. Biol. Chem. 2009, 284, 10013–10022. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Mesaros, A.C.; Blair, I.A.; Penning, T.M. Stereospecific reduction of 5β-reduced steroids by human ketosteroid reductases of the AKR (aldo-keto reductase) superfamily: Role of AKR1C1-AKR1C4 in the metabolism of testosterone and progesterone via the 5β-reductase pathway. Biochem. J. 2011, 437, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Abedin, Z.; Sen, S.; Field, J. Aldo-keto reductases protect lung adenocarcinoma cells from the acute toxicity of B[a]P-7,8-trans-dihydrodiol. Chem. Res. Toxicol. 2012, 25, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.W.; Ho, I.C.; Lee, T.C. Induction of neoplastic transformation by ectopic expression of human aldo-keto reductase 1C isoforms in NIH3T3 cells. Carcinogenesis 2009, 30, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.H.; Chiou, S.H.; Chow, K.C.; Lin, T.Y.; Chang, H.W.; Chiang, I.P.; Lee, M.C. Overexpression of aldo-keto reductase 1C2 is associated with disease progression in patients with prostatic cancer. Histopathology 2010, 57, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Le Calve, B.; Rynkowski, M.; Le Mercier, M.; Bruyere, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Byrns, M.C. Steroid hormone transforming aldo-keto reductases and cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, G.; Ricard, M.J.; Ferris, B.; Strulovici-Barel, Y.; Salit, J.; Hackett, N.R.; Gudas, L.J.; Crystal, R.G. Smoking-induced upregulation of AKR1B10 expression in the airway epithelium of healthy individuals. Chest 2010, 138, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Anttila, S.; Raunio, H.; Hakkola, J. Cytochrome P450-mediated pulmonary metabolism of carcinogens: Regulation and cross-talk in lung carcinogenesis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Palackal, N.T.; Lee, S.H.; Harvey, R.G.; Blair, I.A.; Penning, T.M. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J. Biol. Chem. 2002, 277, 24799–24808. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Higby, R.; Tian, D.; Tan, D.; Johnson, M.D.; Xiao, Y.; Kellar, K.J.; Feng, S.; Shields, P.G. Toxicological analysis of low-nicotine and nicotine-free cigarettes. Toxicology 2008, 249, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Massarsky, A.; Jayasundara, N.; Bailey, J.M.; Oliveri, A.N.; Levin, E.D.; Prasad, G.L.; Di Giulio, R.T. Teratogenic, bioenergetic, and behavioral effects of exposure to total particulate matter on early development of zebrafish (Danio rerio) are not mimicked by nicotine. Neurotoxicol. Teratol. 2015, 51, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Khariwala, S.S.; Hatsukami, D.; Hecht, S.S. Tobacco carcinogen metabolites and DNA adducts as biomarkers in head and neck cancer: Potential screening tools and prognostic indicators. Head Neck 2012, 34, 441–447. [Google Scholar] [CrossRef] [PubMed]
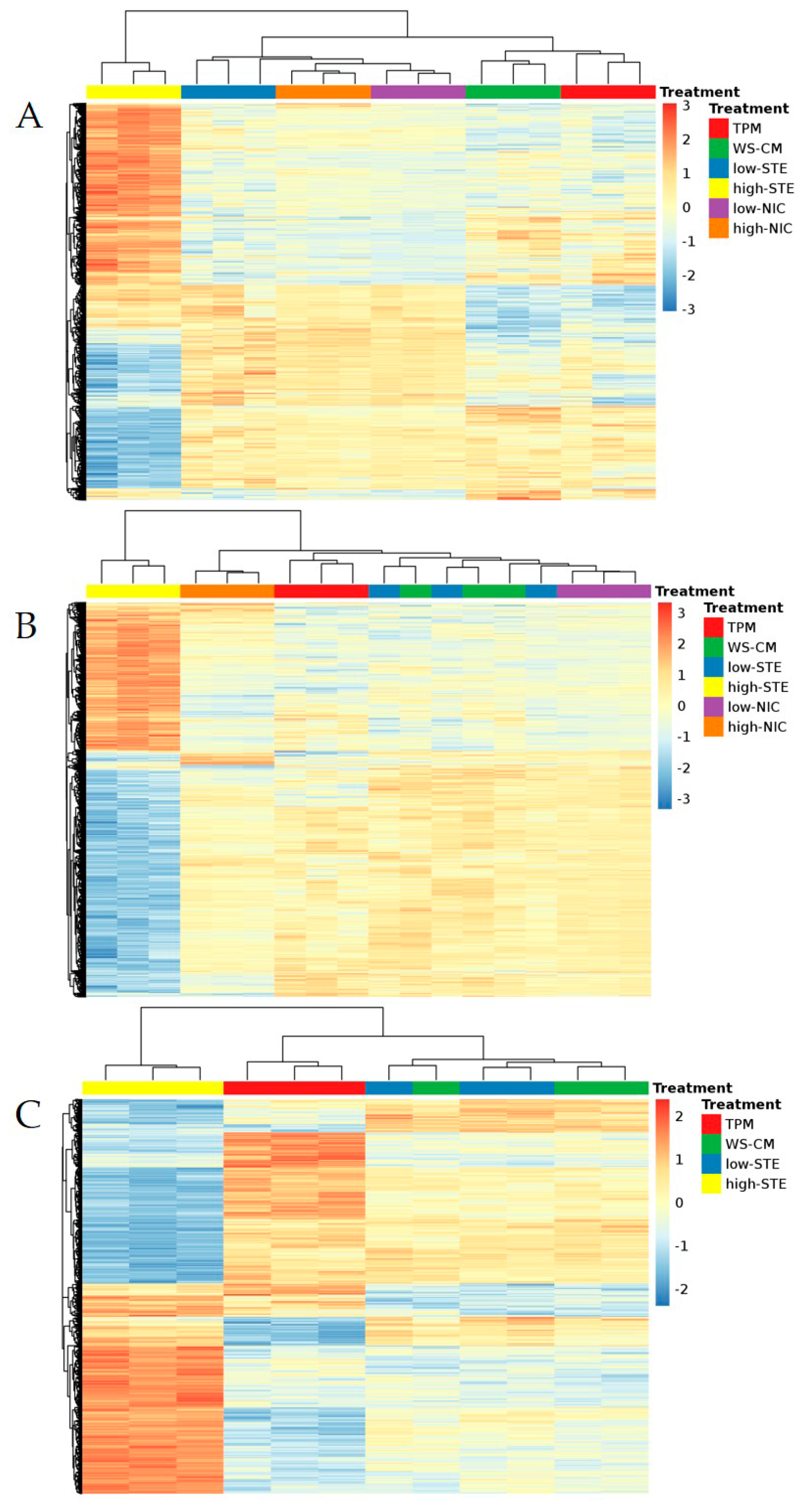

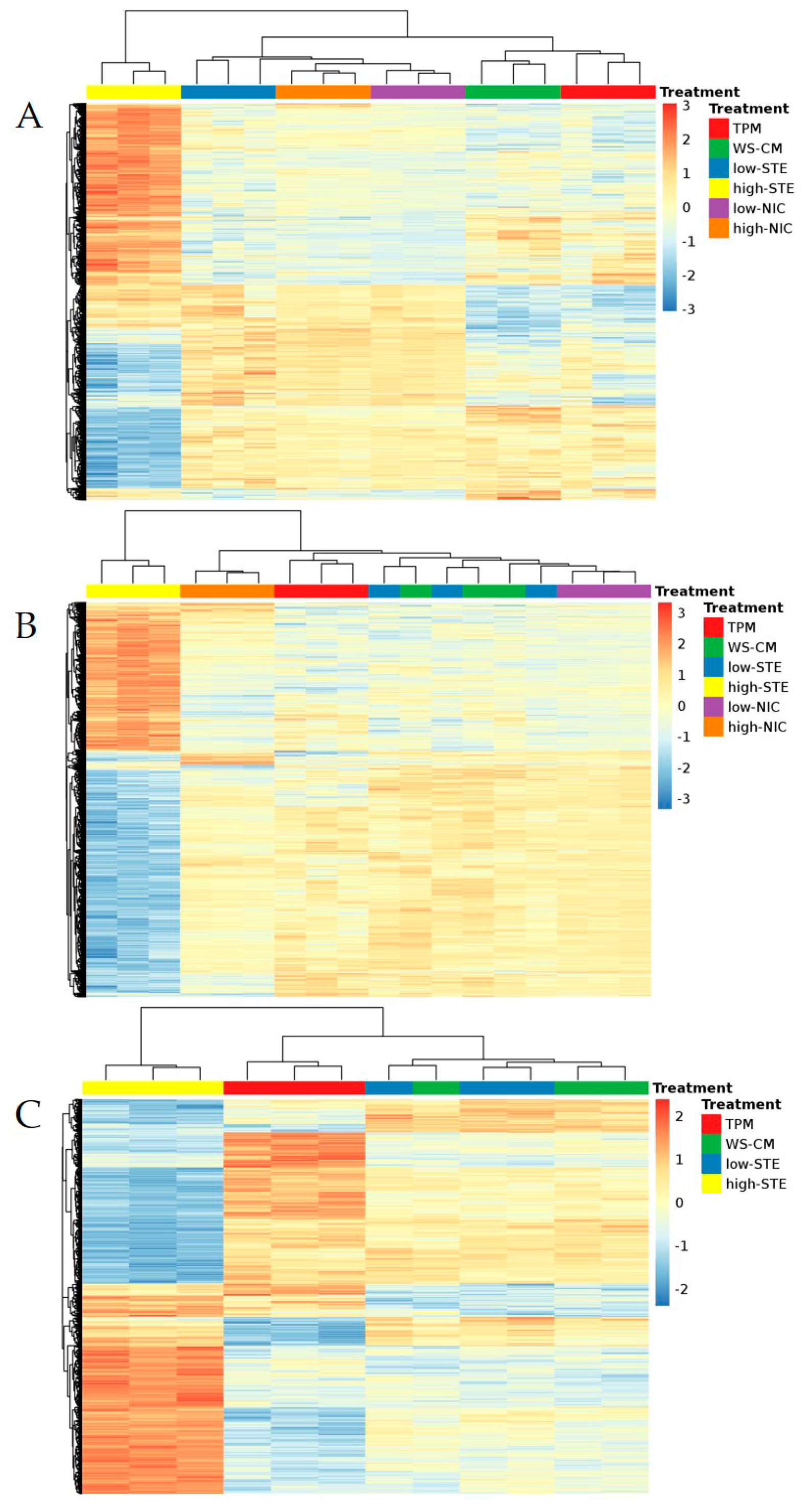{kind=link}
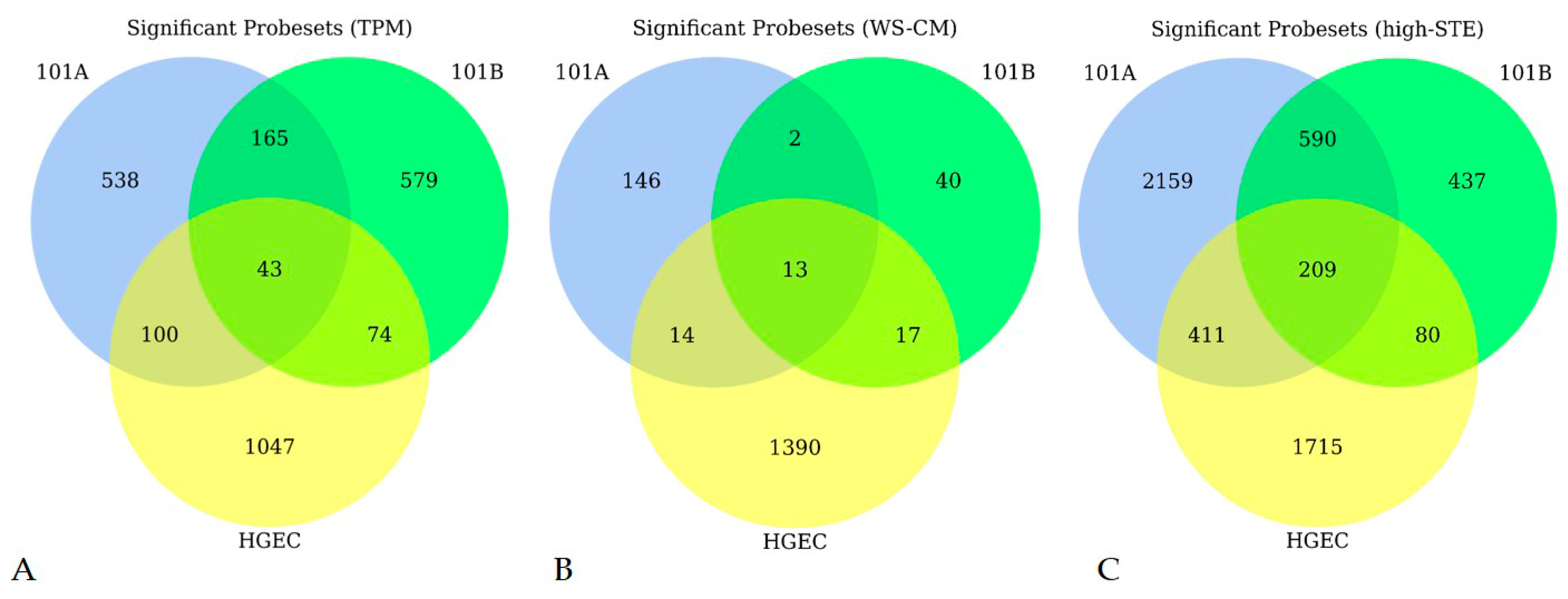{kind=link}
| 101A | 101B | HGEC | |
|---|---|---|---|
| TPM (EC-30) vs. DMSO | 80 µg/mL | 150 µg/mL | 20 µg/mL |
| 9.6 µg/mL NIC | 18 µg/mL NIC | 2.4 µg/mL NIC | |
| Low-STE (containing same amount of NIC as those in EC-30 of TPM) vs. CAS | 148-fold dilution | 79-fold dilution | 592-fold dilution |
| 0.07% (w/v) | 0.13% (w/v) | 0.02% (w/v) | |
| 9.6 µg/mL NIC | 18 µg/mL NIC | 2.4 µg/mL NIC | |
| WS-CM (EC-30) vs. Media | 14-fold dilution | 12.5-fold dilution | 6-fold dilution |
| 1.43% (v/v) | 1.60% (v/v) | 3.33% (v/v) | |
| 1.5 µg/mL NIC | 1.7 µg/mL NIC | 9.3 µg/mL NIC | |
| High-STE vs. CAS | 3-fold dilution | 3-fold dilution | 3-fold dilution |
| 0.33% (w/v) | 0.33% (w/v) | 0.33% (w/v) | |
| 474 µg/mL NIC | 474 µg/mL NIC | 474 µg/mL NIC | |
| low-NIC vs. DMSO | 14 µg/mL NIC | N.D. * | 14 µg/mL NIC |
| high-NIC vs. DMSO | 474 µg/mL NIC | N.D. * | 474 µg/mL NIC |
| Affy ID | Gene Name | Log2 FC | Adjusted p-Values | ||
|---|---|---|---|---|---|
| TPM | WS-CM | TPM | WS-CM | ||
| 1562102_at | 3.45 | 4.96 | 4.4591 × 10−7 | 3.0208 × 10−15 | |
| 1555854_at | AKR1C1, AKR1C2 | 3.12 | 3.52 | 1.5291 × 10−7 | 2.9126 × 10−8 |
| 216594_x_at | AKR1C1, AKR1C2 | 2.37 | 3.01 | 1.4115 × 10−10 | 1.9914 × 10−7 |
| 204151_x_at | AKR1C1, AKR1C2 | 2.38 | 2.65 | 5.6067 × 10−12 | 9.951 × 10−8 |
| 211653_x_at | AKR1C1, AKR1C2 | 2.28 | 2.75 | 4.4159 × 10−10 | 7.8189 × 10−7 |
| 209699_x_at | AKR1C1, AKR1C2 | 2.01 | 2.90 | 1.8013 × 10−9 | 3.075 × 10−7 |
| 203665_at | HMOX1 | 3.87 | 4.72 | 1.7717 × 10−10 | 8.392 × 10−12 |
| 207528_s_at | SLC7A11 | 3.21 | 3.31 | 1.0043 × 10−9 | 6.3878 × 10−10 |
| 217678_at | SLC7A11 | 2.96 | 2.59 | 8.7581 × 10−6 | 2.4987 × 10−9 |
| 209921_at | SLC7A11 | 2.94 | 2.55 | 8.7581 × 10−6 | 2.7297 × 10−9 |
| 202831_at | GPX2 | 1.45 | 2.38 | 0.00188523 | 8.0032 × 10−10 |
| 231897_at | PTGR1 | 0.80 | 1.73 | 0.00504145 | 5.9494 × 10−11 |
| KEGG PATHWAY | TPM | WS-CM | Low-STE | High-STE | Low-NIC | High-NIC |
|---|---|---|---|---|---|---|
| q-value (NES) * | q-value (NES) * | q-value (NES) * | q-value (NES) * | q-value (NES) * | q-value (NES) * | |
| METABOLISM_OF_XENOBIOTICS_BY_CYTOCHROME_P450 | 0 (2.41) | 0 (2.37) | -- | 0.0095 (1.90) | -- | -- |
| STEROID_HORMONE_BIOSYNTHESIS | 0 (2.36) | 0.0014 (2.05) | -- | 0.0050 (2.00) | -- | -- |
| RETINOL_METABOLISM | 0.0018 (2.02) | -- | -- | -- | -- | -- |
| PHENYLALANINE_METABOLISM | -- | -- | 0.0475 (−1.73) | -- | -- | -- |
| PORPHYRIN_AND_CHLOROPHYLL_METABOLISM | -- | 0.0616 (1.78) | -- | -- | -- | -- |
| SYSTEMIC_LUPUS_ERYTHEMATOSUS | -- | -- | -- | -- | -- | 0.1501 (−1.72) |
| CELL_CYCLE | 0 (−2.48) | 0 (−2.30) | -- | -- | -- | -- |
| DNA_REPLICATION | 0 (−2.30) | 0 (−2.18) | -- | -- | -- | 0.1628 (−1.67) |
| OOCYTE_MEIOSIS | -- | 0.0007 (−2.05) | -- | -- | -- | -- |
| MISMATCH_REPAIR | 0.0016 (−1.98) | -- | -- | -- | -- | -- |
| GLYCINE_SERINE_AND_THREONINE_METABOLISM | -- | -- | 0.0273 (−1.85) | -- | -- | -- |
| OLFACTORY_TRANSDUCTION | -- | -- | 0.0286 (−1.80) | -- | -- | -- |
| ARACHIDONIC_ACID_METABOLISM | -- | -- | -- | 0.0050 (1.95) | -- | -- |
| CYTOSOLIC_DNA_SENSING_PATHWAY | -- | -- | -- | 0.0186 (−1.93) | -- | -- |
| PEROXISOME | -- | -- | -- | 0.0942 (−1.75) | -- | -- |
| RIG_I_LIKE_RECEPTOR_SIGNALING_PATHWAY | -- | -- | -- | 0.0676 (−1.74) | -- | -- |
| FATTY_ACID_METABOLISM | -- | -- | -- | -- | -- | 0.2090 (−1.75) |
| TPPs | Microarray | qRT-PCR | ||||
|---|---|---|---|---|---|---|
| AKR1C1/AKR1C2 | AKR1C1 | AKR1C2 | ||||
| Fold Change | Adj. p-Value | Fold Change | p-Value | Fold Change | p-Value | |
| TPM | 8.69 | <0.001 | 16.43 | <0.001 | 1.95 | 0.3121 |
| WS-CM | 11.47 | <0.001 | 33.28 | <0.001 | 2.07 | 0.2719 |
| low-STE | 1.08 | 1 | 1.23 | 0.5689 | 1.61 | 0.8227 |
| high-NIC | 1.59 | 0.5413 | 1.61 | 0.2052 | 0.59 | 0.4112 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, S.; Gao, H.; Henderson, D.; Zacharias, W.; Liu, G.; Tran, Q.T.; Prasad, G.L. AKR1C1 as a Biomarker for Differentiating the Biological Effects of Combustible from Non-Combustible Tobacco Products. Genes 2017, 8, 132. https://doi.org/10.3390/genes8050132
Woo S, Gao H, Henderson D, Zacharias W, Liu G, Tran QT, Prasad GL. AKR1C1 as a Biomarker for Differentiating the Biological Effects of Combustible from Non-Combustible Tobacco Products. Genes. 2017; 8(5):132. https://doi.org/10.3390/genes8050132
Chicago/Turabian StyleWoo, Sangsoon, Hong Gao, David Henderson, Wolfgang Zacharias, Gang Liu, Quynh T. Tran, and G.L. Prasad. 2017. "AKR1C1 as a Biomarker for Differentiating the Biological Effects of Combustible from Non-Combustible Tobacco Products" Genes 8, no. 5: 132. https://doi.org/10.3390/genes8050132
APA StyleWoo, S., Gao, H., Henderson, D., Zacharias, W., Liu, G., Tran, Q. T., & Prasad, G. L. (2017). AKR1C1 as a Biomarker for Differentiating the Biological Effects of Combustible from Non-Combustible Tobacco Products. Genes, 8(5), 132. https://doi.org/10.3390/genes8050132