
Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Genome Sequencing, Assembly and Annotation
2.3. Genome Repeat, Gene Selective Pressure Analysis, Phylogenetic Analysis
2.4. Sequence Divergence Analysis
3. Results
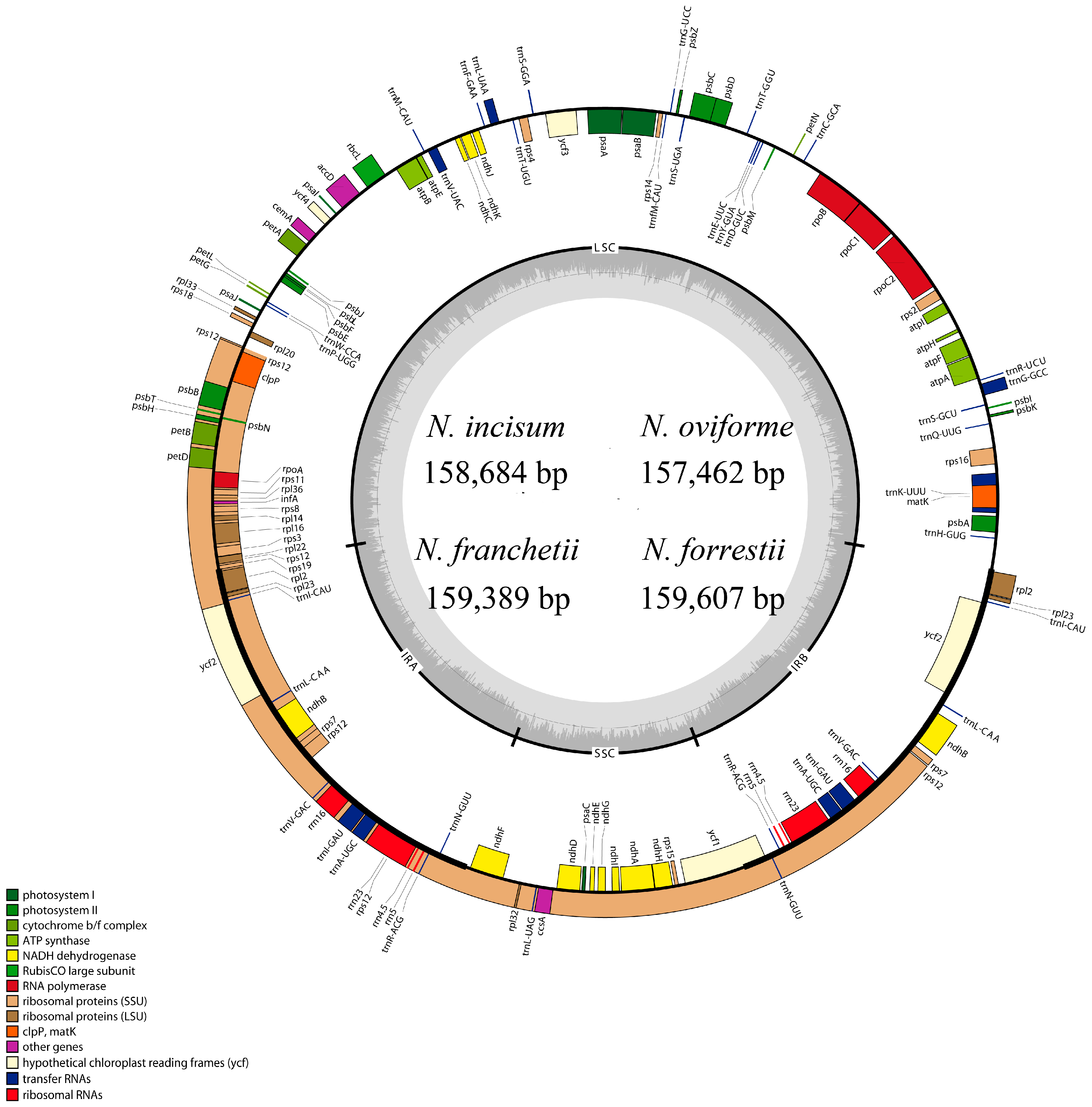
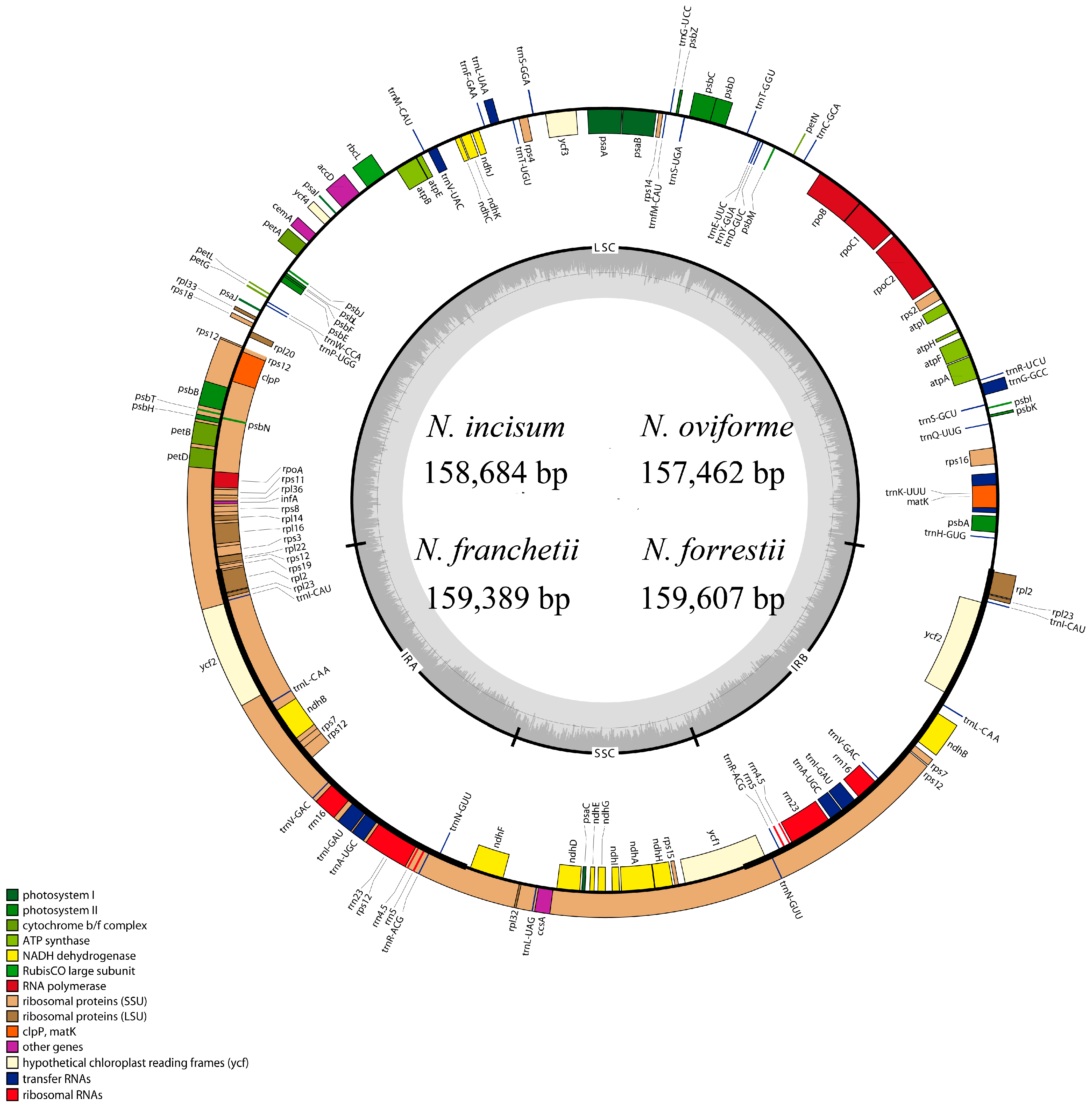
3.1. The Overall Features of cp DNA of Four Notopterygium Species
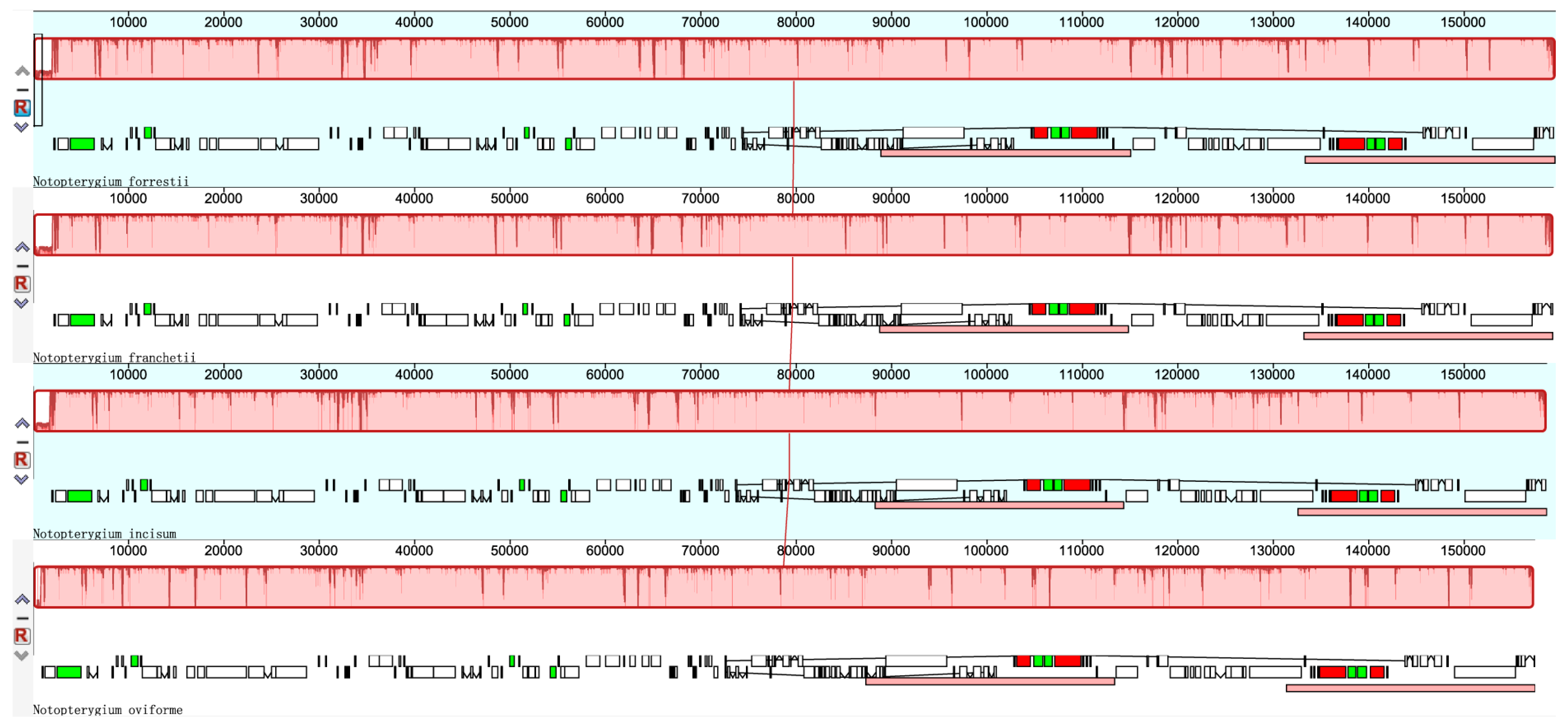
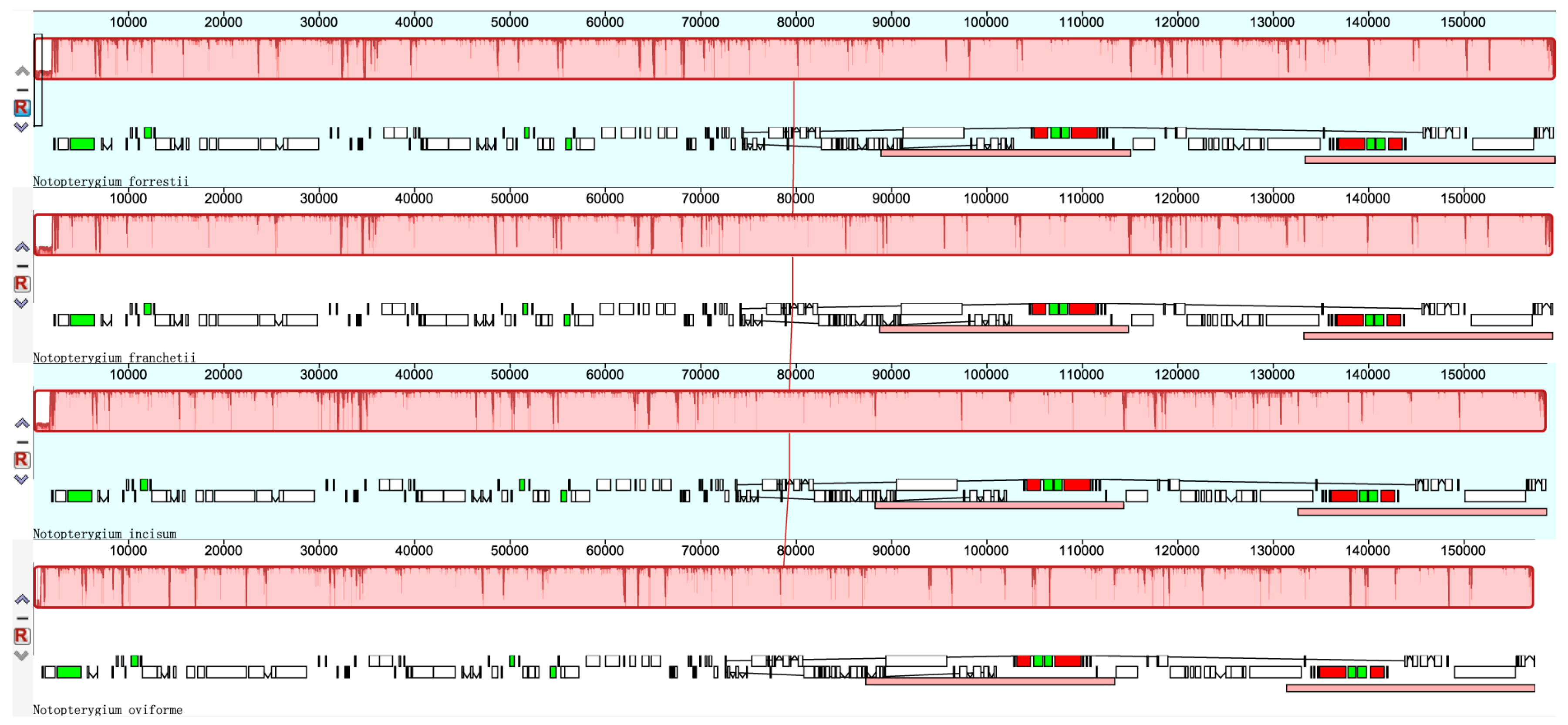
3.2. Comparisons of cp DNA of four Notopterygium Species
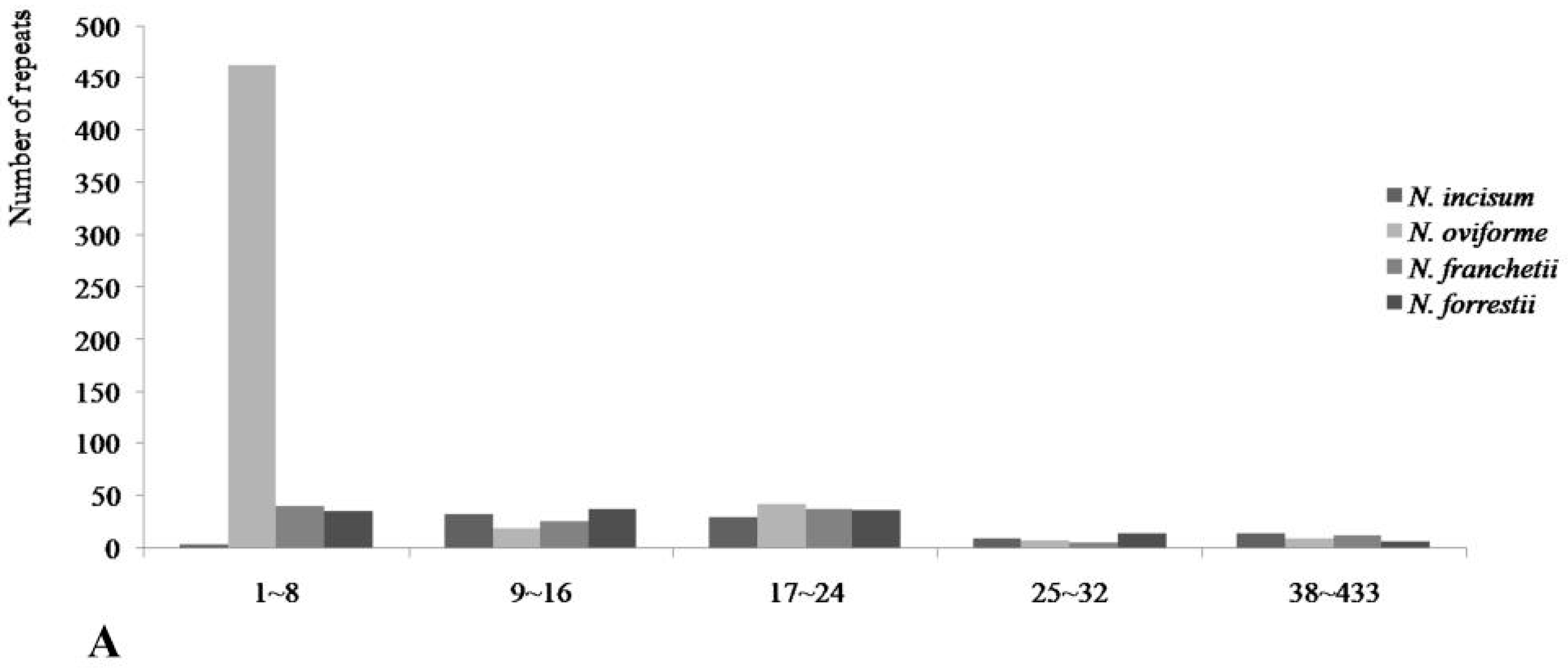
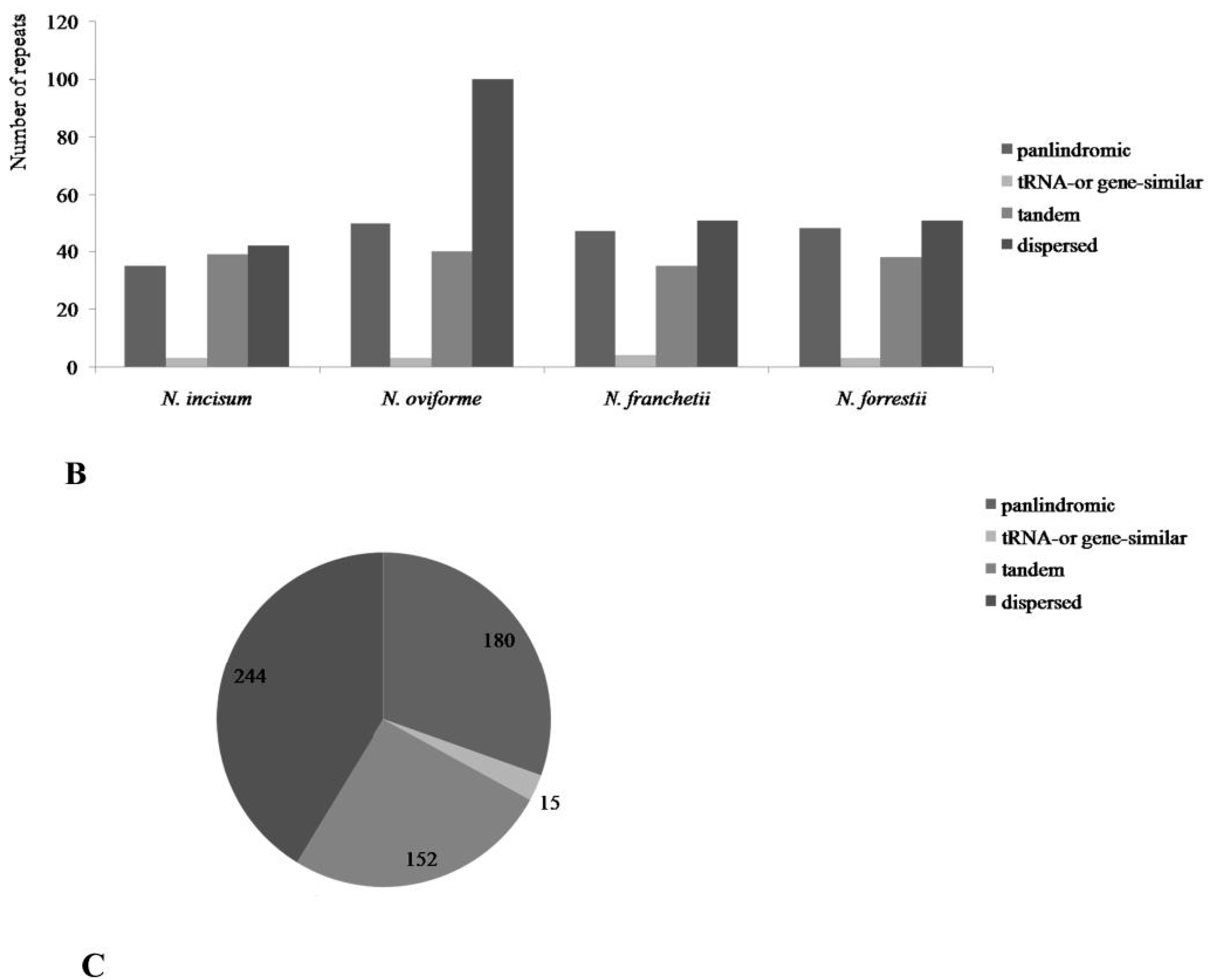
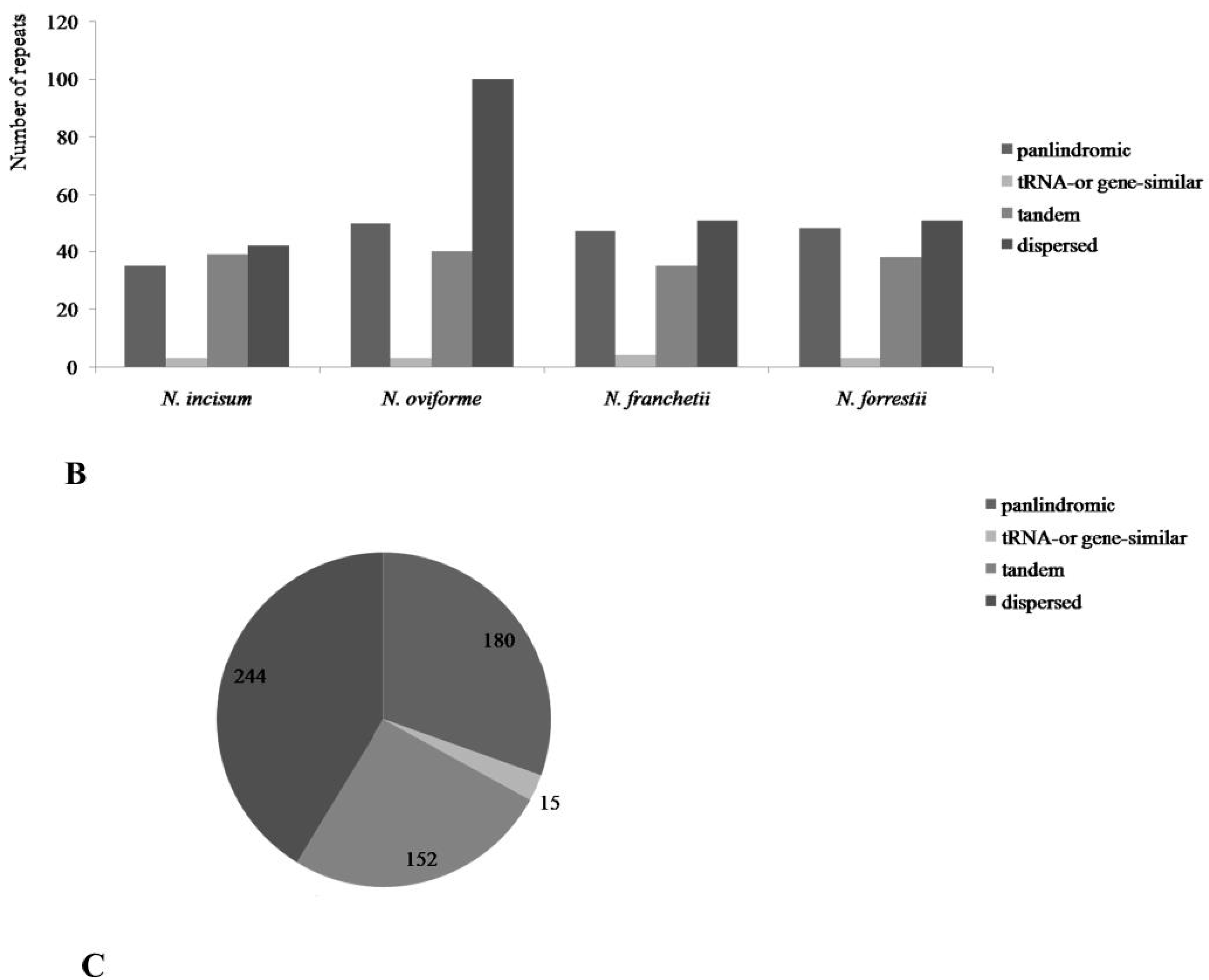
3.3. Repetitive Sequences
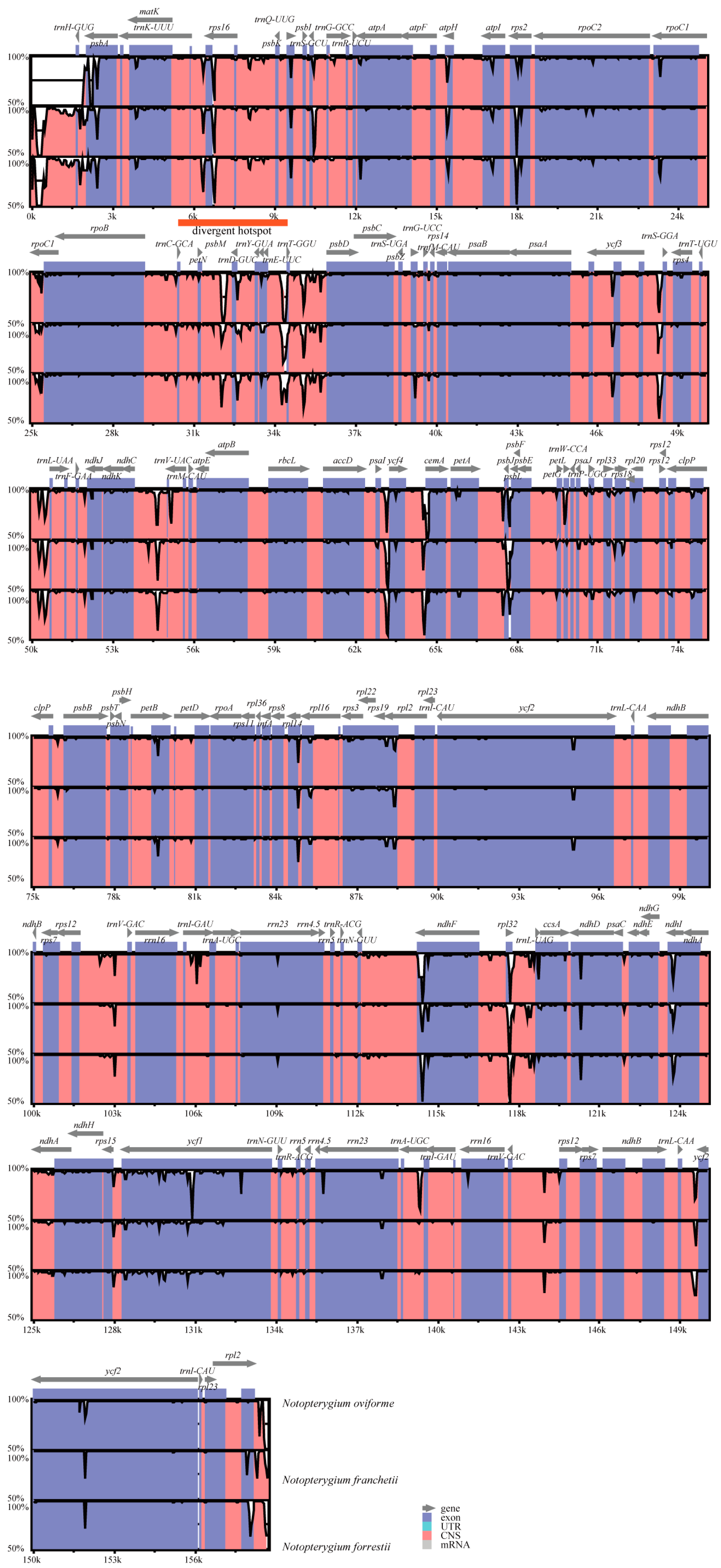
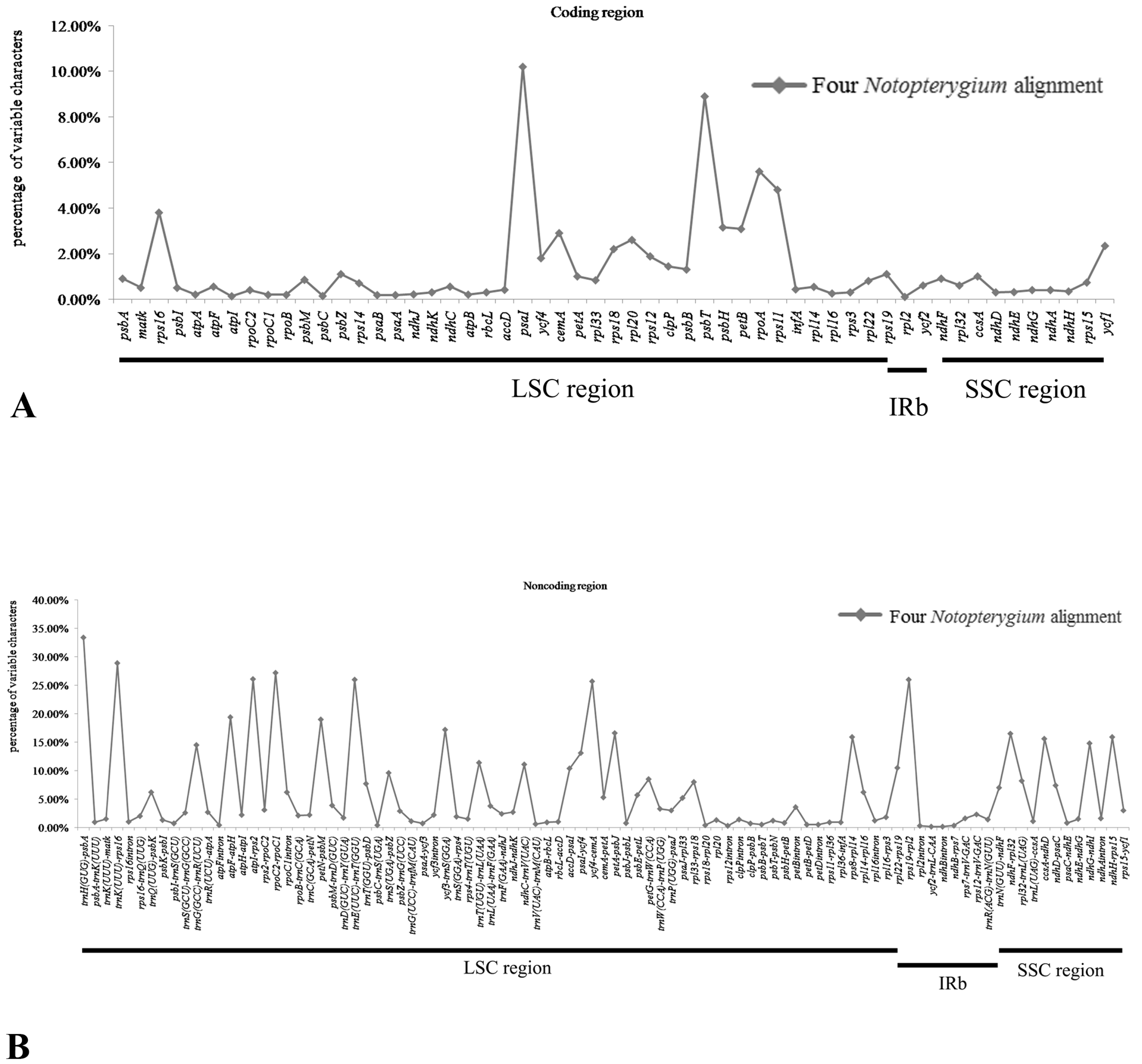
3.4. Sequence Divergence among Notopterygium Species
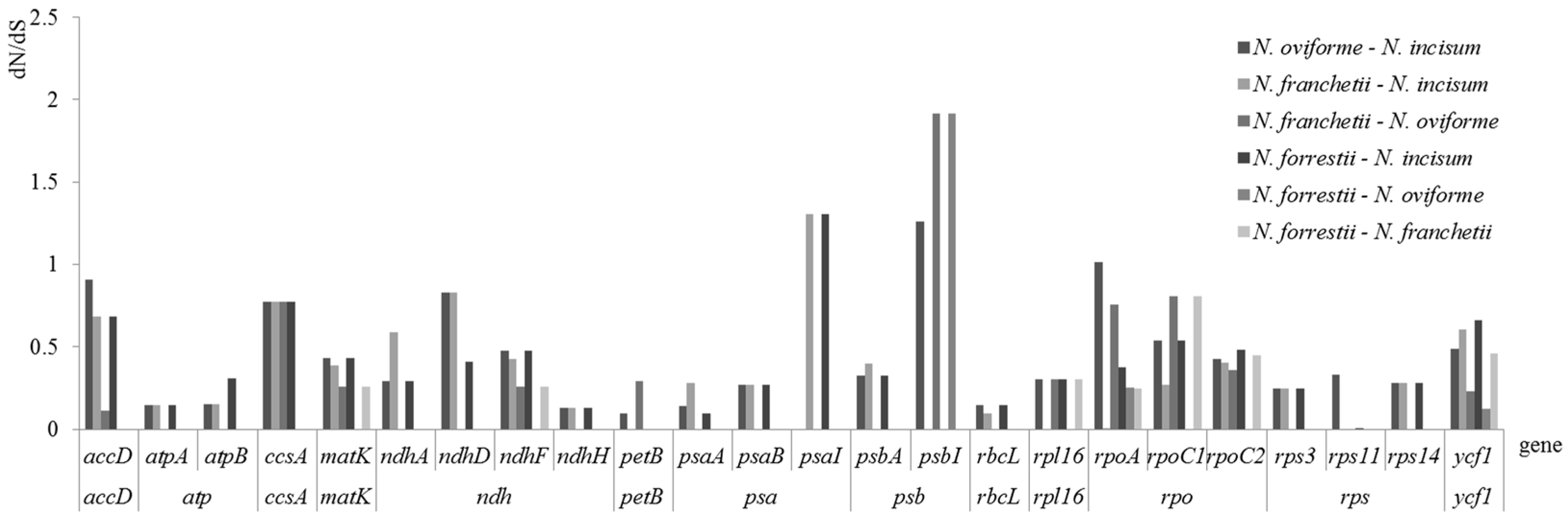
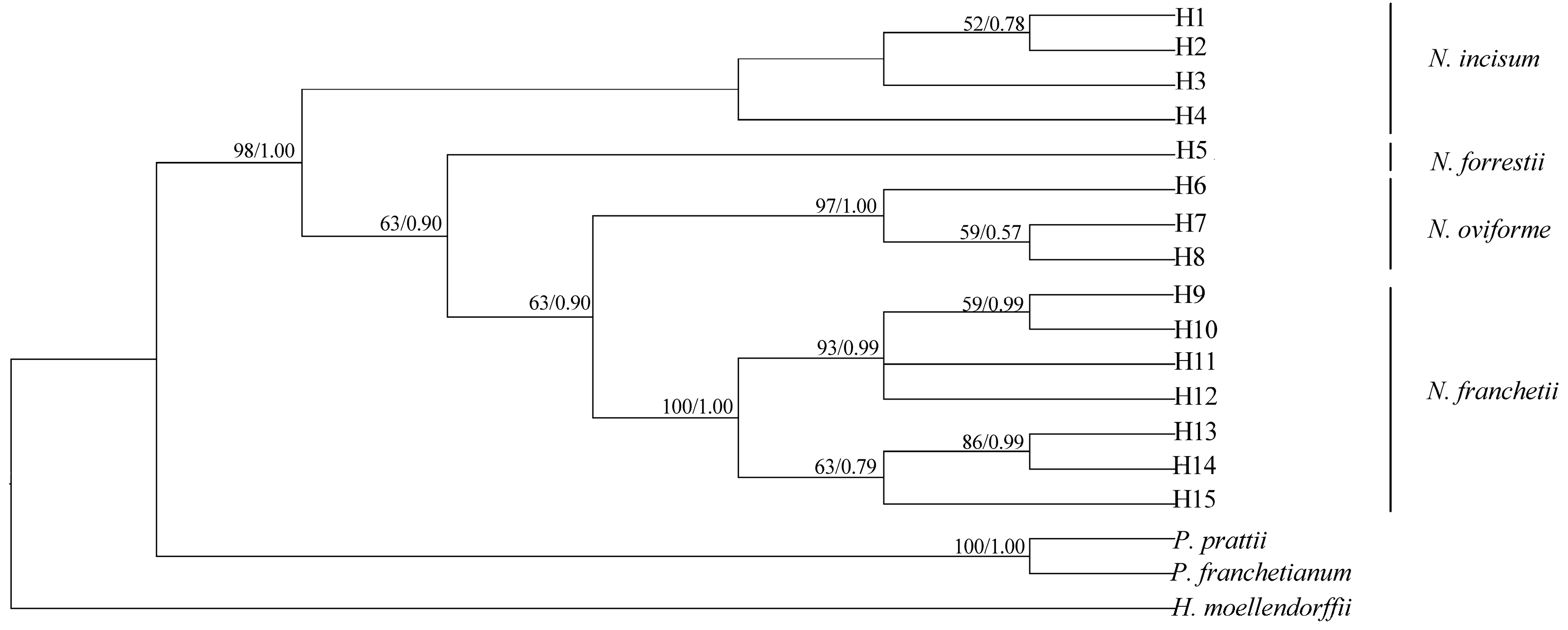
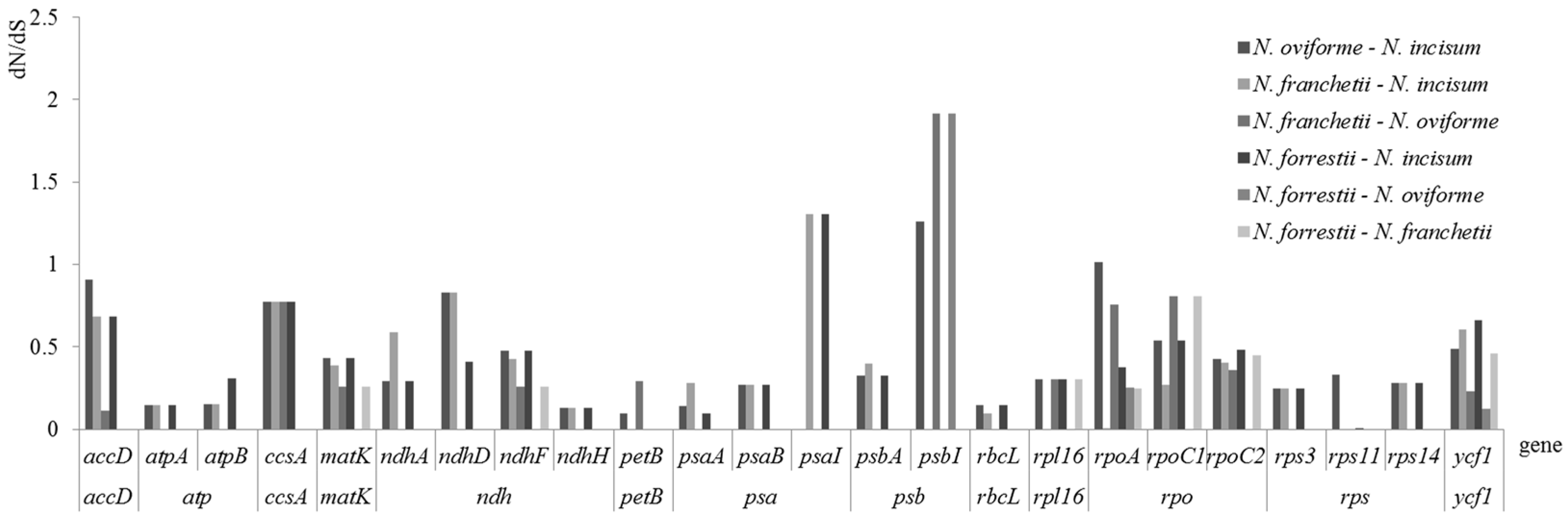
3.5. Evolutionary Rates of Notopterygium
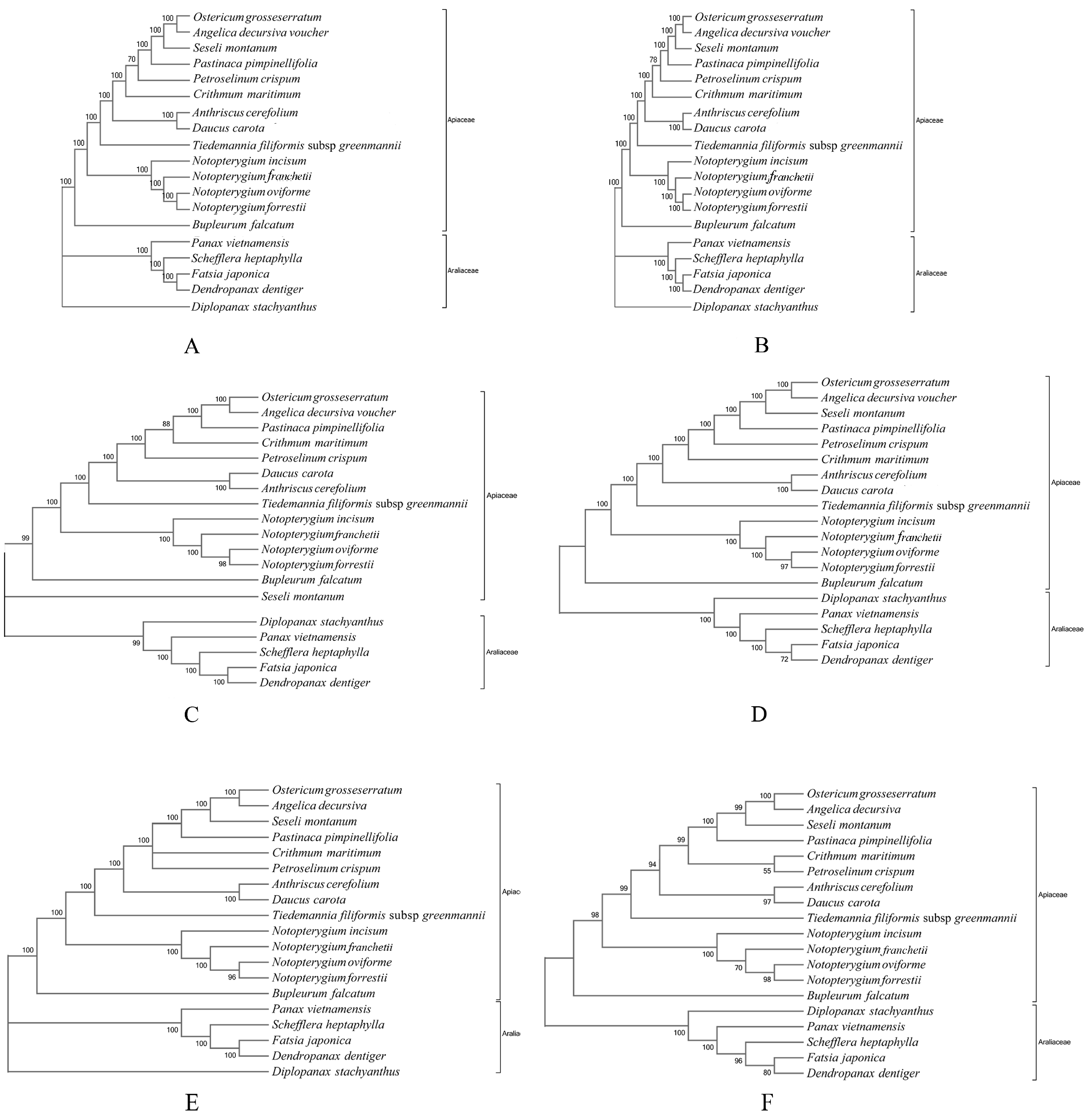
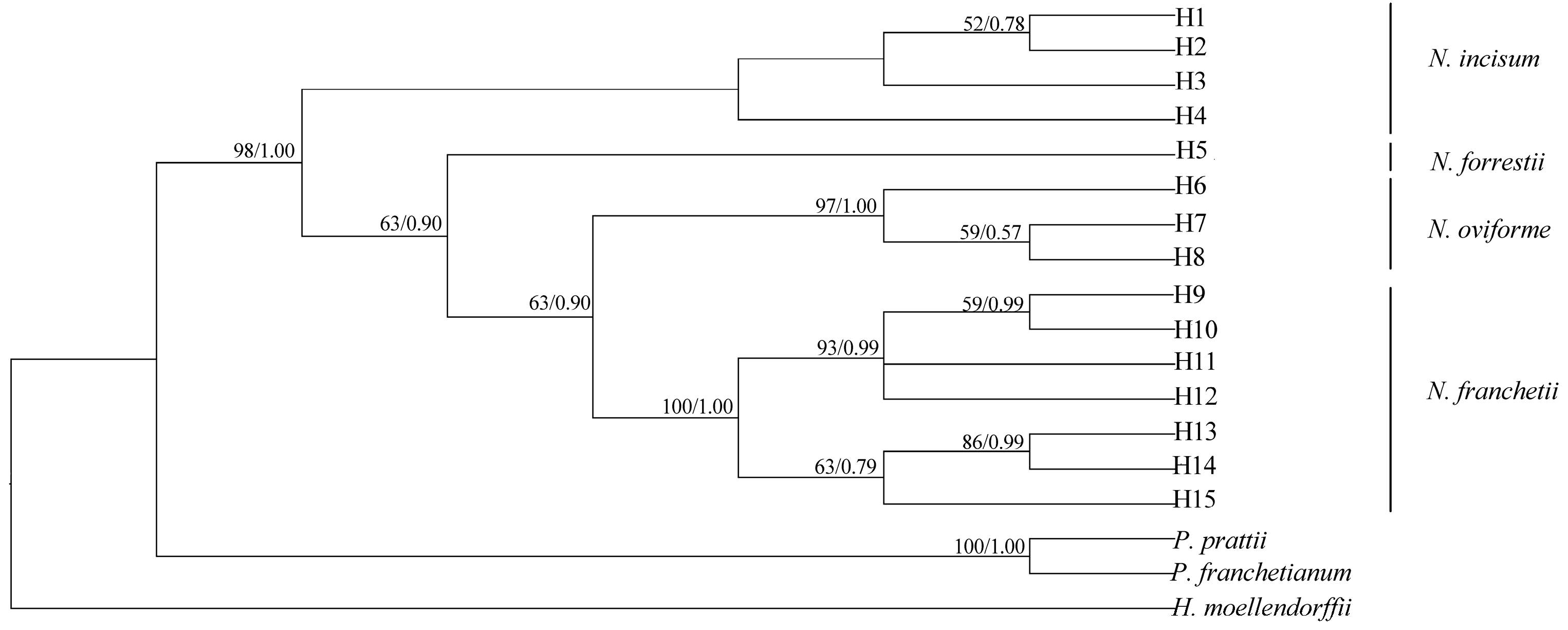
3.6. Phylogenomic Relationship of Notopterygium Species
4. Discussion
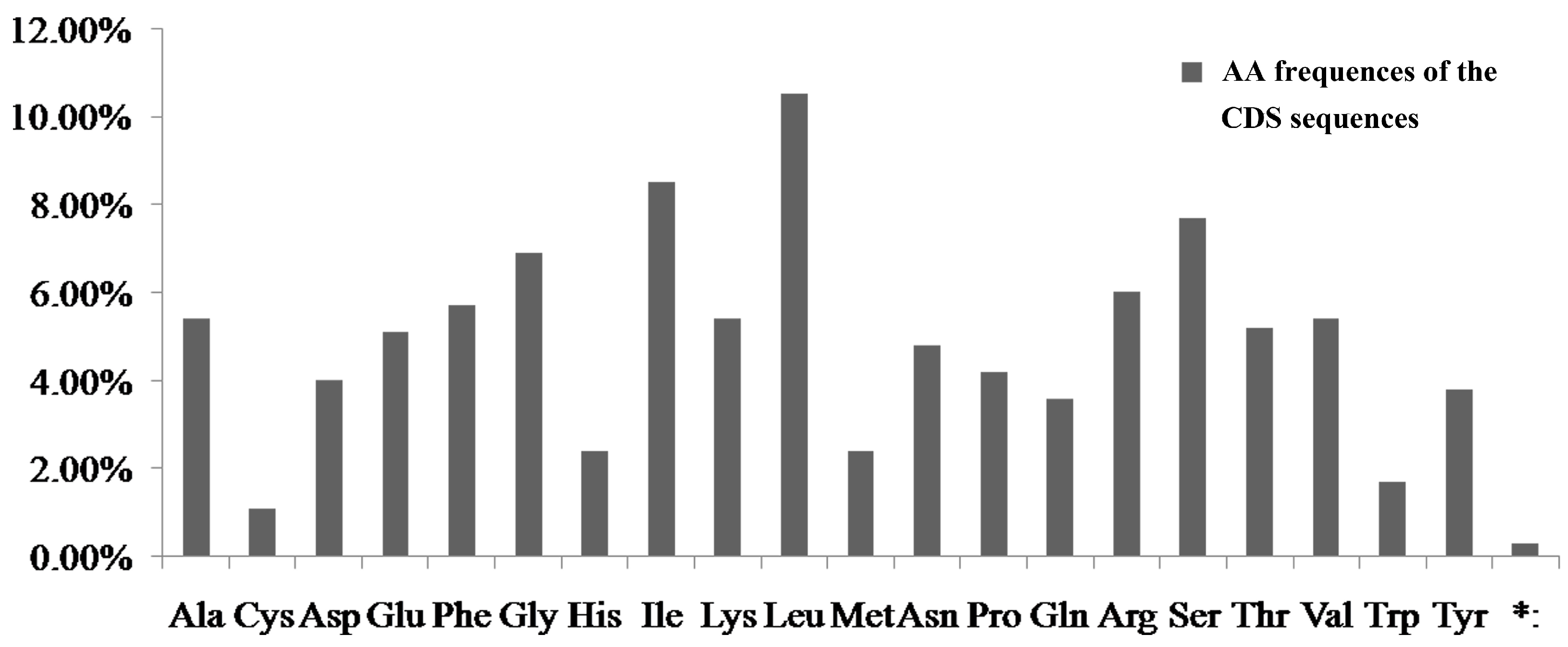
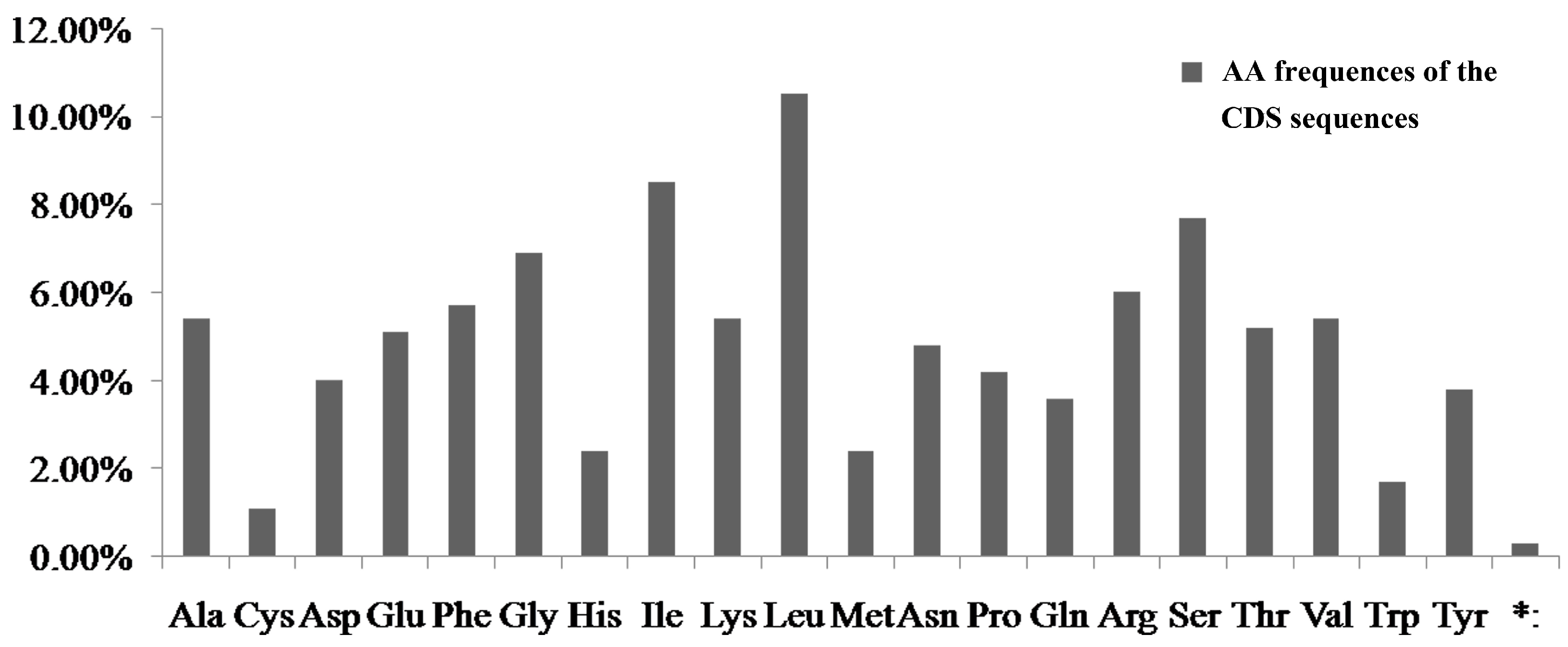
4.1. The Relationship of AT Content and Codon Usage of the Four Notopterygium Species
4.2. Sequence Divergence
4.3. Phylogenomic Relationships among the Four Notopterygium Species
4.4. Evolutionary Rates among Notopterygium Species
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, Z.Y.; Raven, P.H. Flora of China. Volume 14. Apiaceae through Ericaceae; Science Press: Beijing, China, 2005. [Google Scholar]
- Xu, H.B.; Sun, X.B. Study on the pharmacological effects of volatile oil of Notopterygium incisum. Chin. Tradit. Herb. Drugs 1991, 22, 28–30. [Google Scholar]
- Zhou, Y.; Jiang, S.Y.; Ma, X.J.; Sun, H.; Pu, F.D.; Wu, R. Resource crisis and protective measures on rhizoma et radix Notopterygii. Chin. Tradit. Herb. Drugs 2003, 34, 12–14. [Google Scholar]
- Wang, Y.P.; Pu, F.D.; Wang, P.L.; He, X.J. Studies on the systematics of the Chinese endemic genus Notopterygium. Acta Bot. Yunnanica 1995, 18, 424–430. [Google Scholar]
- Pu, F.D.; Wang, P.L.; Zheng, Z.H.; Wang, Y.P. A reclassification of Notopterygium Boissieu (Umbelliferae). Acta. Phytotaxon. Sin. 1999, 38, 430–436. [Google Scholar]
- Jiang, S.Y.; Sun, H.; Huang, X.J.; Zhou, Y.; Ma, X.J.; Yang, Z.R. Environmental pedology of Notopterygium incisum and N. forbesii. Chin. Tradit. Herb. Drugs 2005, 36, 918–921. [Google Scholar]
- Yang, X.W.; Zhang, P.; Tao, H.Y.; Jiang, S.Y.; Zhou, Y. GC-MS analysis of essential oil constituents from rhizome and root of Notopterygium forbesii. J. Chin. Pharm. Sci. 2006, 15, 200–205. [Google Scholar]
- Wang, S. China Species Red List; Higher Education Press: Beijing, China, 2004. [Google Scholar]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, C.R.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The chloroplast genome. Plant Mol. Biol. 1992, 19, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The chloroplast genome. Essays Biochem. 1994, 30, 49–57. [Google Scholar]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2009, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Palmer, J.D. A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proc. Natl. Acad. Sci. USA 1987, 84, 5818–5822. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Plastid chromosomes: Structure and evolution. In Molecular Biology of Plastids; Bogorad, L., Vasil, I.K., Eds.; Academic Press: San Diego, CA, USA, 1991; pp. 5–53. [Google Scholar]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two new world bamboos-Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)-extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.F.; Zanis, M.J.; Emery, N.C. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 2014, 101, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Ané, C.; Burleigh, J.G.; McMahon, M.M.; Sanderson, M.J. Covarion structure in plastid genome evolution: A new statistical test. Mol. Biol. Evol. 2005, 22, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Huang, J.P.; Wu, C.S.; Hsu, C.Y.; Chaw, S.M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Bio. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F.; Davis, J.I.; Leebens-Mack, J.; Conran, J.G.; Stevenson, D.W. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 2013, 29, 65–87. [Google Scholar] [CrossRef]
- Raman, G.; Choi, K.S.; Park, S. Phylogenetic relationships of the fern Cyrtomium falcatum (Dryopteridaceae) from Dokdo island based on chloroplast genome sequencing. Genes 2016, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.K.; Yan, H.D.; Zhao, X.X.; Zhang, X.Q.; Wang, J.; Frazier, T.; Yin, G.; Huang, X.; Yan, D.F.; Zang, W.J.; et al. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol. Ecol. Resour. 2015, 15, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, Y.F.; Deng, C.; Ma, Y.; Wang, Z.W.; Chen, X.H.; Xue, L.B. Comparative transcriptome analysis of eggplant (Solanum melongena L.) and turkey berry (Solanum torvum Sw.): Phylogenomics and disease resistance analysis. BMC Genom. 2014, 15, 412. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Zhao, S.Y.; Wang, Q.F.; Moody, M.L. Transcriptome sequencing of three Ranunculus species (Ranunculaceae) reveals candidate genes in adaptation from terrestrial to aquatic habitats. Sci. Rep. 2014, 5, 10098. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, R.; Huang, L.; Zheng, X.M.; Liu, P.L.; Du, Y.S.; Cai, Z.; Zhou, L.; Wei, X.H.; Zhang, F.M.; et al. Widespread and adaptive alterations in genome-wide gene expression associated with ecological divergence of two Oryza species. Mol. Biol. Evol. 2016, 33, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Patel, R.K.; Jain, M. NGSQCToolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [PubMed]
- Li, W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Pamilo, P.; Bianchi, N. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evol. 1993, 10, 271–281. [Google Scholar] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.L.; Lan, S.R.; Cai, B.P.; Chen, S.P.; Chen, H.; Zhou, S.L. The complete chloroplast genome of Guadua angustifolia and comparative analyses of neotropical-paleotropical Bamboos. PLoS ONE 2015, 10, e0143792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony and other Methods, version 4.0; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, w273–w279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Song, J.Y.; Gao, H.H.; Zhu, Y.J.; Xu, J.; Pang, X.H.; Yao, H.; Sun, C.; Li, X.E.; Li, C.Y.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [PubMed]
- Morton, B.R. Chloroplast DNA codon use: Evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 1993, 37, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Li, W.H. The rate of synonymous substitution in enterobacterial genes is inversely related to codon usage bias. Mol. Biol. Evol. 1987, 4, 222–230. [Google Scholar] [PubMed]
- Kwon, K.C.; Chan, H.T.; León, I.R.; Williams-Carrier, R.; Barkan, A.; Daniell, H. Codon-optimization to enhance expression yields insights into chloroplast translation. Plant Physiol. 2016, 27, 00981. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G.; Bernardi, G. Compositional constraints and genome evolution. J. Mol. Evol. 1986, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Morton, B.R. Codon adaptation of plastid genes. PLoS ONE 2016, 11, e0154306. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Tsudzuki, T.; Takahashi, S.; Shimada, H.; Kadowaki, K.I. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: A comparative analysis of four monocot chloroplast genomes. DNA Res. 2004, 11, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Davis, J.I.; Soreng, R.J.; Garvin, D.; Anderson, M.J. Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc. Natl. Acad. Sci. USA 1992, 89, 7722–7726. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.; Hu, J.M.; Kuo, C.H. Complete plastid genome sequence of the basal asterid Ardisia polysticta Miq. and comparative analyses of asterid plastid genomes. PLoS ONE 2013, 8, e62548. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kössel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, K.; Hodkinson, T.R.; Wolfe, K.H.; van den Bekerom, R.; Dix, P.J.; Barth, S. Complete chloroplast genome sequence of a major allogamous forage species, perennial ryegrass (Lolium perenne L.). DNA Res. 2009, 16, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; dePamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.F.; Zhang, Y.X.; Zeng, C.X.; Guo, Z.H.; Li, D.Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Reboud, X.; Zeyl, C. Organelle inheritance in plants. Heredity 1994, 72, 132–140. [Google Scholar] [CrossRef]
- Feng, J.J.; Jiang, D.C.; Shang, H.Y.; Dong, M.; Wang, G.N.; He, X.Y.; Zhao, C.; Mao, K. Barcoding poplars (Populus L.) from western China. PLoS ONE 2013, 8, e71710. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.C.; Premoli, A.C. Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenet. Evol. 2010, 54, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pelser, P.B.; Kennedy, A.H.; Tepe, E.J.; Shidler, J.B.; Nordenstam, B.; Kadereit, J.W.; Watson, L.E. Patterns and causes of incongruence between plastid and nuclear Senecioneae (Asteraceae) phylogenies. Am. J. Bot. 2010, 97, 856–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Phylogenetic and taxonomic status analyses of the abaso section in populus from multiple nuclear genes and plastid fragments reveal new insights into the North America origin of Populus. Front. Plant Sci. 2016, 7, 2022. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef]
- Hu, S.; Sablok, G.; Wang, B.; Qu, D.; Barbaro, E.; Viola, R.; Li, M.A.; Varotto, C. Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 2015, 16, 306. [Google Scholar] [CrossRef] [PubMed]
- Ometto, L.; Li, M.; Bresadola, L.; Varotto, C. Rates of evolution in stress-related genes are associated with habitat preference in two Cardamine lineages. BMC Evol. Biol. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, S.I.; Nagano, A.J.; Miyazaki, S.; Kubo, M.; Demura, T.; Fukuda, H.; Sakai, S.; Hasebe, M. Ecogenomics of cleistogamous and chasmogamous flowering: Genome-wide gene expression patterns from cross-species microarray analysis in Cardamine kokaiensis (Brassicaceae). J. Ecol. 2008, 96, 1086–1097. [Google Scholar] [CrossRef]
- Streb, P.; Shang, W.; Feierabend, J.; Bligny, R. Divergent strategies of photoprotection in high-mountain plants. Planta 1998, 207, 313–324. [Google Scholar] [CrossRef]
- Germino, M.J.; Smith, W.K. High resistance to low-temperature photo inhibition in two alpine, snowbank species. Physiol. Plant. 2000, 110, 89–95. [Google Scholar] [CrossRef]
- Frohnmeyer, H.; Staiger, D. Ultraviolet-B radiation-mediated responses in plants. Balancing damage and protection. Plant Physiol. 2003, 133, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Streb, P.; Aubert, S.; Gout, E.; Bligny, R. Cold and light induced changes of metabolite and antioxidant levels in two high mountain plant species Soldanella alpina and Ranunculus glacialis and a lowland species Pisum sativum. Physiol. Plant. 2003, 118, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Fujii, N.; Setoguchi, H. Molecular evolution of phytochromes in Cardamine nipponica (Brassicaceae) suggests the involvement of PHYE in local adaptation. Genetics 2009, 182, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Tomoko, O. Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 1995, 40, 56–63. [Google Scholar] [CrossRef]
- McInerney, J.O. The causes of protein evolutionary rate variation. Trends Ecol. Evol. 2006, 21, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, C.; Kobayashi, Y.; Aoki, S.; Sugita, C.; Sugita, M. Complete chloroplast DNA sequence of the moss Physcomitrella patens: Evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 2003, 31, 5324–5331. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Madoka, Y.; Tomizawa, K.I.; Mizoi, J.; Nishida, I.; Nagano, Y.; Sasaki, Y. Chloroplast transformation with modified accD operon increases acetyl-CoA carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiol. 2002, 43, 1518–1525. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Size (bp) | LSC (bp) | SSC (bp) | IR (bp) | Number of Protein-Coding Genes a | Number of tRNA Genes a | Number of rRNA Genes a | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| N. incisum | 158,684 | 88,260 | 18,232 | 26,096 | 85 (6) | 37 (7) | 8 (4) | 37.70 |
| N. oviforme | 157,462 | 87,304 | 17,996 | 26,081 | 85 (6) | 37 (7) | 8 (4) | 37.90 |
| N. franchetii | 159,389 | 88,749 | 18,290 | 26,175 | 85 (6) | 37 (7) | 8 (4) | 37.70 |
| N. forrestii | 159,607 | 88,870 | 18,212 | 26,262 | 85 (6) | 37 (7) | 8 (4) | 37.70 |
| Category | Gene Group | Gene Name | ||||
|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn16 | rrn23 | rrn4.5 | rrn5 | |
| Transfer RNA genes | trnI-CAU(2) | trnI-GAU(2) | trnL-UAA | trnL-CAA(2) | trnL-UAG | |
| trnR-UCU | trnR-ACG(2) | trnA-UGC(2) | trnW-CCA | trnM-CAU | ||
| trnV-UAC | trnV-GAC(2) | trnF-GAA | trnT-UGU | trnT-GGU | ||
| trnP-UGG | trnfM-CAU | trnG-UCC | trnG-GCC | trnS-GGA | ||
| trnS-UGA | trnS-GCU | trnD-GUC | trnC-GCA | trnN-GUU(2) | ||
| trnE-UUC | trnY-GUA | trnQ-UUG | trnK-UUU | trnH-GUG | ||
| Small subunit of ribosome | rps2 | rps3 | rps4 | rps7 | rps8 | |
| rps11 | rps12 | rps14 | rps15 | rps16 | ||
| rps18 | rps19 | |||||
| Large subunit of ribosome | rp12 | rp114 | rp116 | rp120 | rp122 | |
| rp123 | rp132 | rp133 | rp136 | |||
| DNA-dependent RNA polymerase | rpoA | rpoB | rpoC1 | rpoC2 | ||
| Translational initiation factor | infA | |||||
| Genes for photosynthesis | Subunits of photosystem I | psaA | psaB | psaC | psaI | psaJ |
| ycf3 | ycf4 | |||||
| Subunits of photosystem II | psbA | psbB | psbC | psbD | psbE | |
| psbF | psbH | psbI | psbJ | psbK | ||
| psbL | psbM | psbN | psbT | psbZ | ||
| NADH oxidoreductase | ndhA | ndhB | ndhC | ndhD | ndhE | |
| ndhF | ndhG | ndhH | ndhI | ndhJ | ||
| ndhK | ||||||
| Subunits of cytochrome | petA | petB | petD | petG | petL | |
| petN | ||||||
| Subunits of ATP | atpA | atpB | atpE | atpF | atpH | |
| synthase | atpI | |||||
| Large subunit of Rubisco | rbcL | |||||
| Other genes | Maturase | matk | ||||
| Envelope membrane protein | cemA | |||||
| Subunit of acetyl-CoA | accD | |||||
| C-type cytochrome synthesis gene | ccsA | |||||
| Protease | clpP | |||||
| Conserved reading frames | Conserved Open Reading Frames | ycf1 | ycf2 | |||
| Region | N. incisum | N. oviforme | N. franchetii | N. forrestii | |
|---|---|---|---|---|---|
| LSC (%) | A | 31.5 | 31.4 | 31.5 | 31.6 |
| T | 32.7 | 32.6 | 32.7 | 32.6 | |
| C | 18.4 | 18.3 | 18.4 | 18.4 | |
| G | 17.4 | 17.7 | 17.4 | 17.4 | |
| GC | 35.8 | 36.0 | 35.8 | 35.8 | |
| SSC (%) | A | 34.6 | 34.4 | 34.6 | 34.6 |
| T | 33.8 | 33.8 | 33.8 | 33.8 | |
| C | 16.5 | 16.5 | 16.5 | 16.4 | |
| G | 15.1 | 15.2 | 15.1 | 15.1 | |
| GC | 31.6 | 31.7 | 31.6 | 31.6 | |
| IRa (%) | A | 28.3 | 28.2 | 28.3 | 28.3 |
| T | 28.7 | 28.5 | 28.7 | 28.7 | |
| C | 22.1 | 22.1 | 22.2 | 22.2 | |
| G | 20.8 | 21.2 | 20.8 | 20.8 | |
| GC | 42.9 | 43.3 | 43.0 | 43.0 | |
| IRb (%) | A | 28.7 | 28.6 | 28.7 | 28.6 |
| T | 28.3 | 28.2 | 28.3 | 28.4 | |
| C | 20.8 | 21.0 | 20.8 | 20.8 | |
| G | 22.1 | 22.2 | 22.2 | 22.2 | |
| GC | 42.9 | 43.2 | 43.0 | 43.0 | |
| overall length (%) | A | 30.9 | 30.8 | 30.9 | 30.9 |
| T | 31.4 | 31.3 | 31.4 | 31.4 | |
| C | 19.2 | 19.2 | 19.2 | 19.2 | |
| G | 18.5 | 18.8 | 18.5 | 18.5 | |
| GC | 37.7 | 37.9 | 37.7 | 37.7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Yue, M.; Niu, C.; Ma, X.-F.; Li, Z.-H. Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes 2017, 8, 124. https://doi.org/10.3390/genes8040124
Yang J, Yue M, Niu C, Ma X-F, Li Z-H. Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes. 2017; 8(4):124. https://doi.org/10.3390/genes8040124
Chicago/Turabian StyleYang, Jiao, Ming Yue, Chuan Niu, Xiong-Feng Ma, and Zhong-Hu Li. 2017. "Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium" Genes 8, no. 4: 124. https://doi.org/10.3390/genes8040124
APA StyleYang, J., Yue, M., Niu, C., Ma, X.-F., & Li, Z.-H. (2017). Comparative Analysis of the Complete Chloroplast Genome of Four Endangered Herbals of Notopterygium. Genes, 8(4), 124. https://doi.org/10.3390/genes8040124