Homoeologous Recombination of the V1r1-V1r2 Gene Cluster of Pheromone Receptors in an Allotetraploid Lineage of Teleosts


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.1.1. Sample Collection and Evaluation of Polyploidy
2.1.2. Amplification, Cloning and Sequencing
2.2. Data Analysis
2.2.1. Sequence Characterization and Alignment and Phylogenetic Reconstruction
2.2.2. Recombination and Selection Analyses
3. Results and Discussion
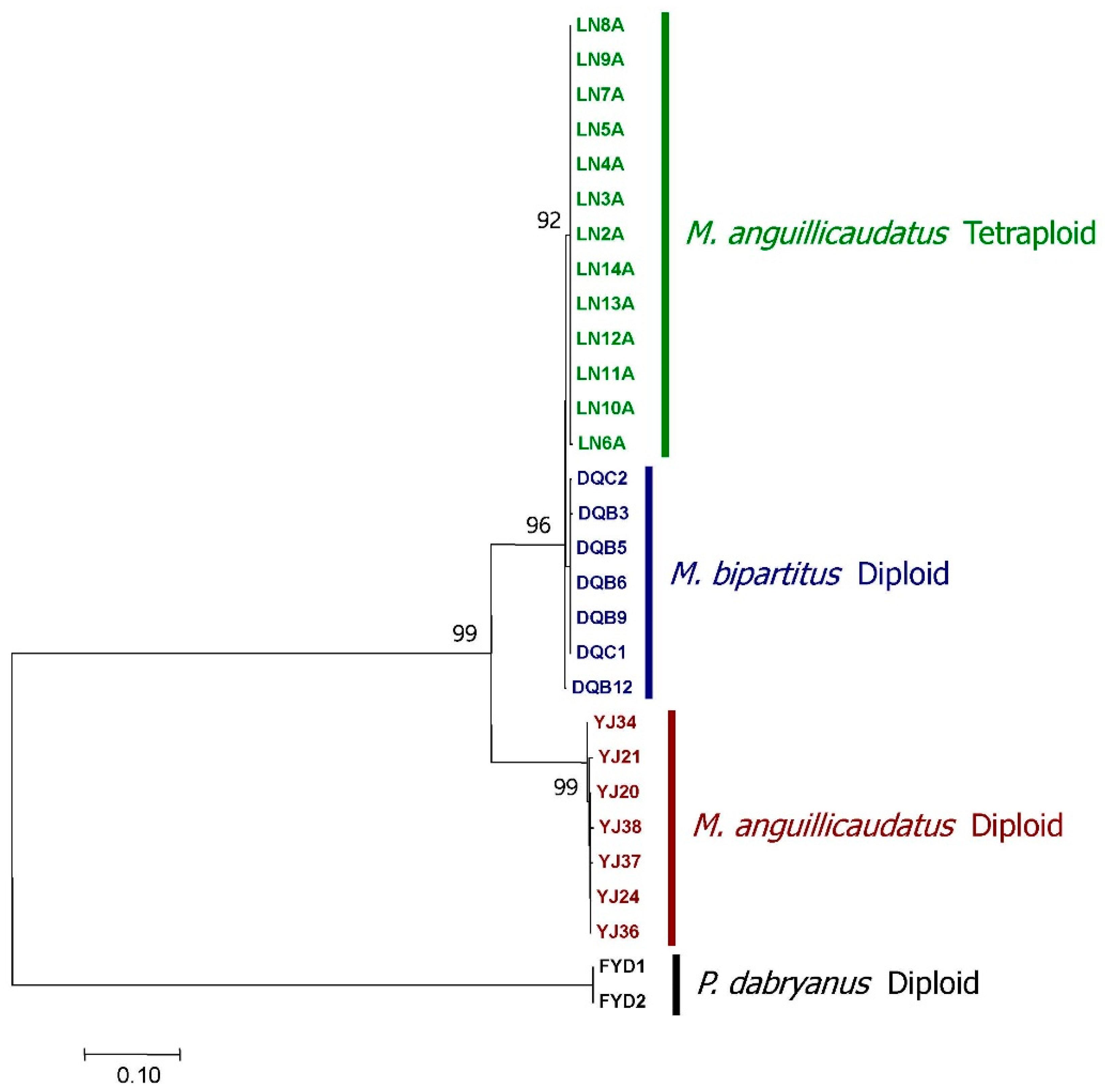
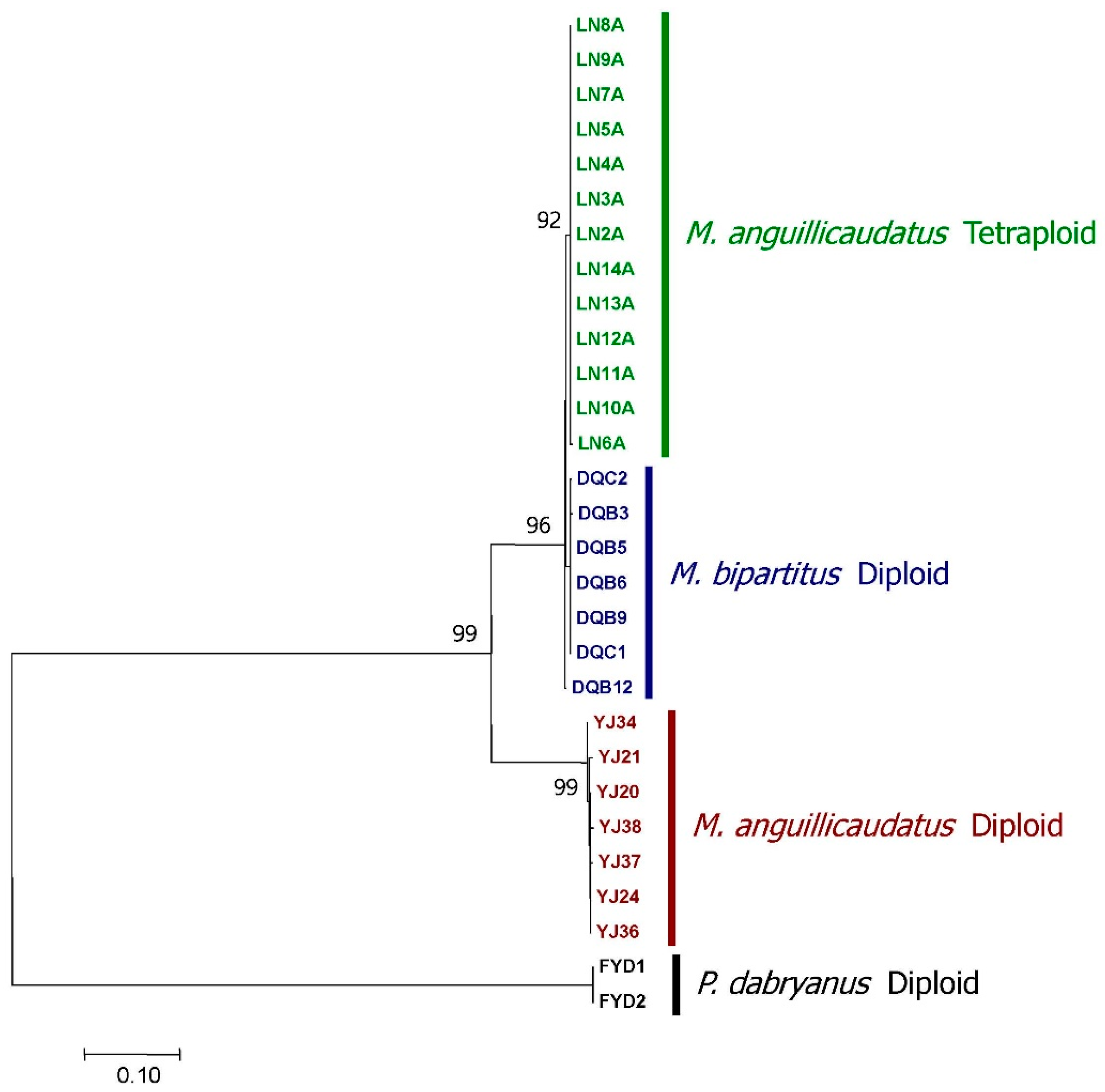
3.1. Identification of an Allotetraploid Lineage of Oriental Weatherfishes
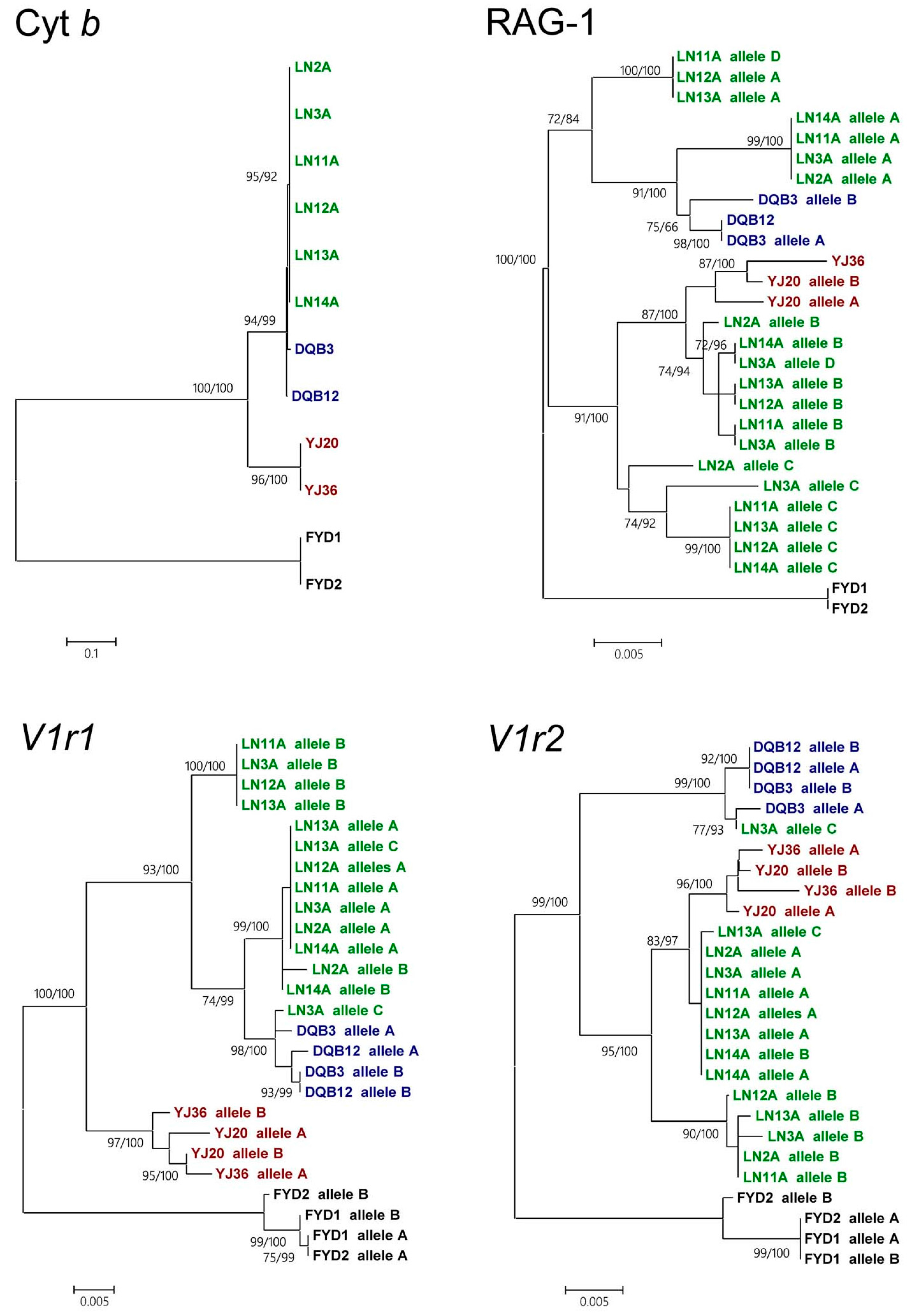
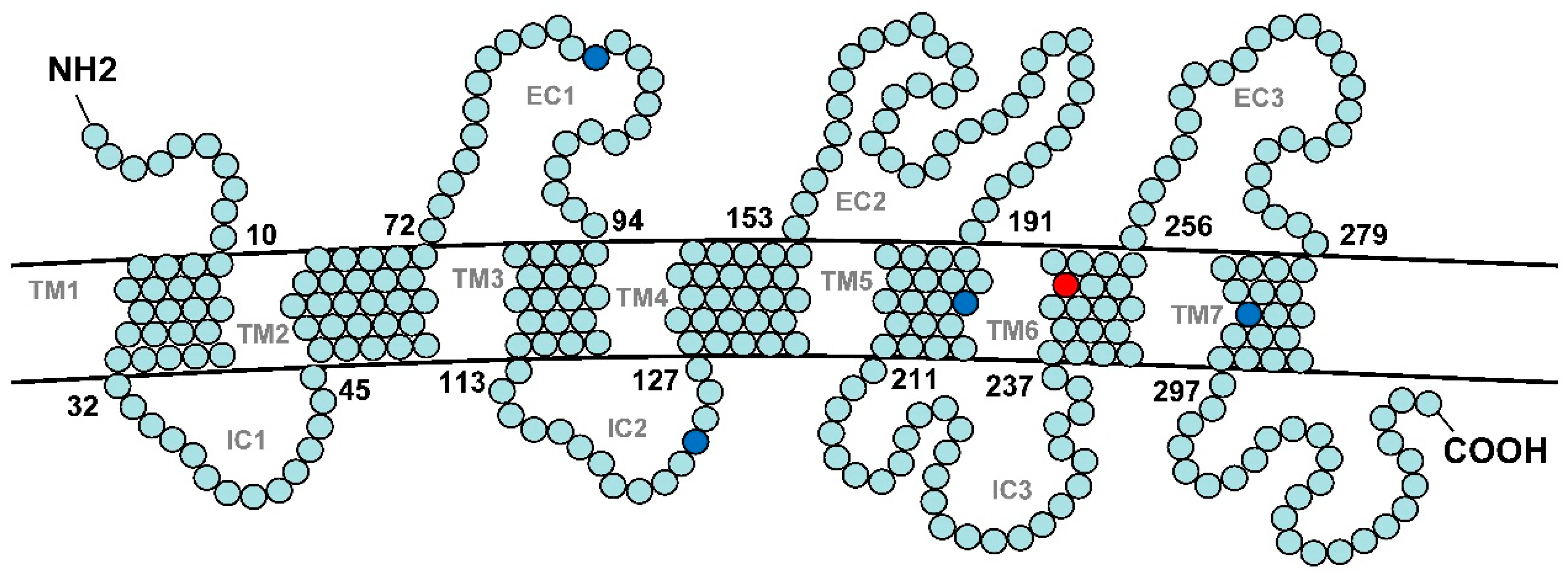
3.2. Characterization of the V1r1 and V1r2 Genes of Weatherfishes
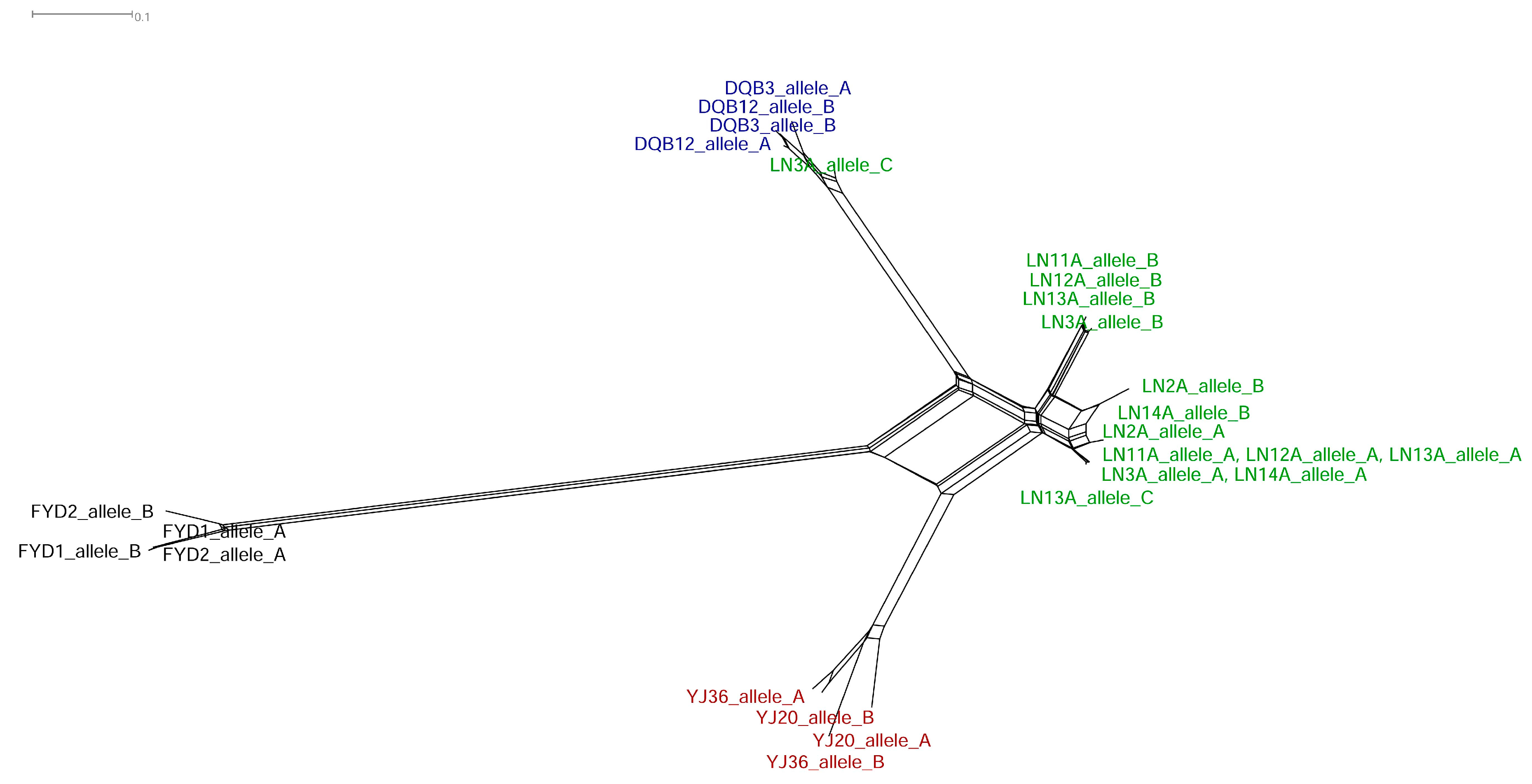
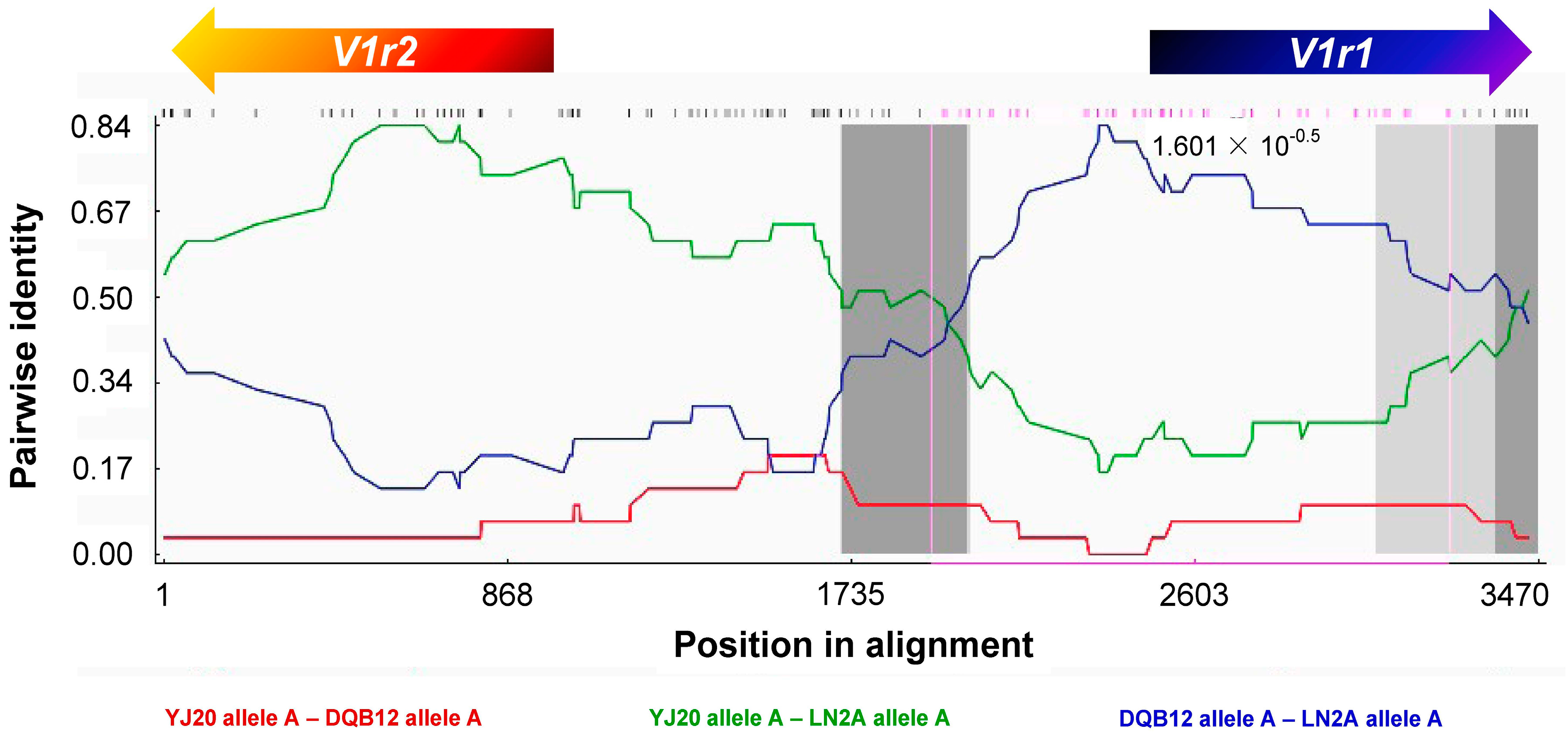
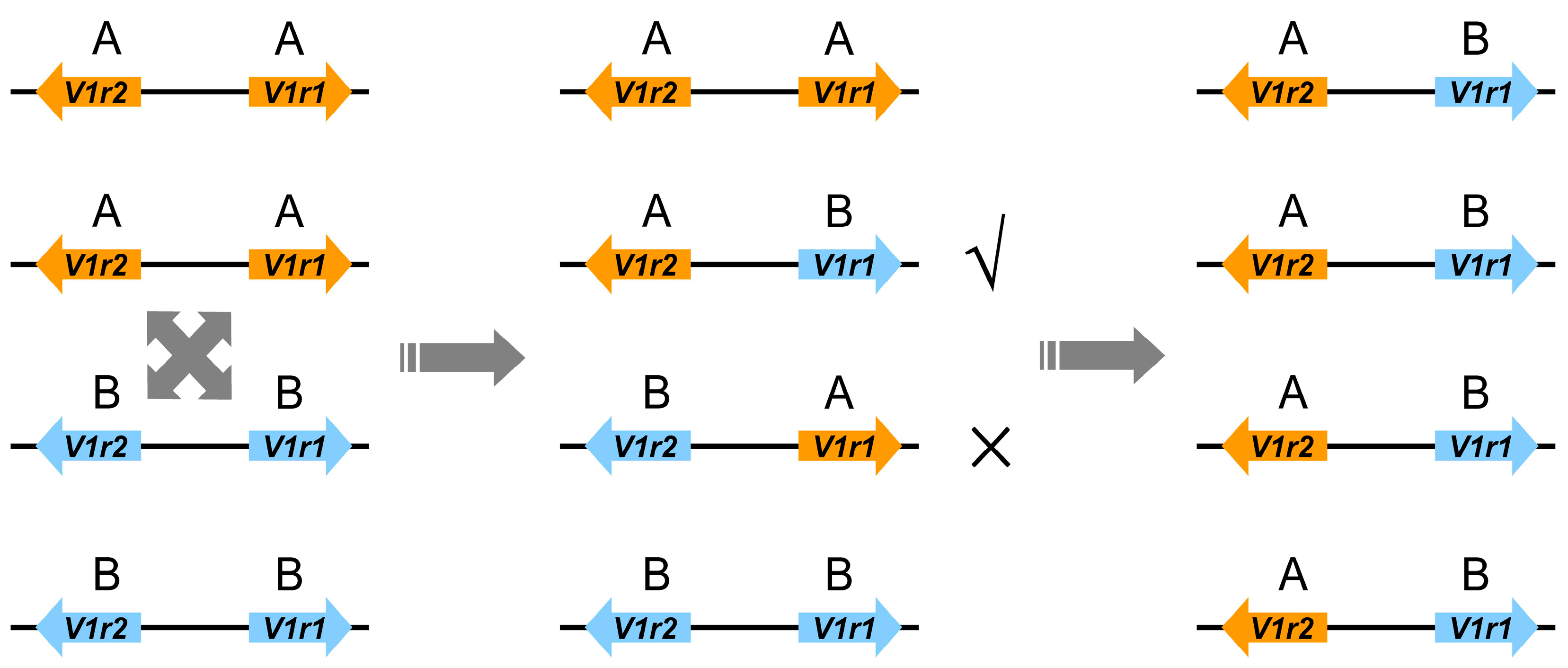
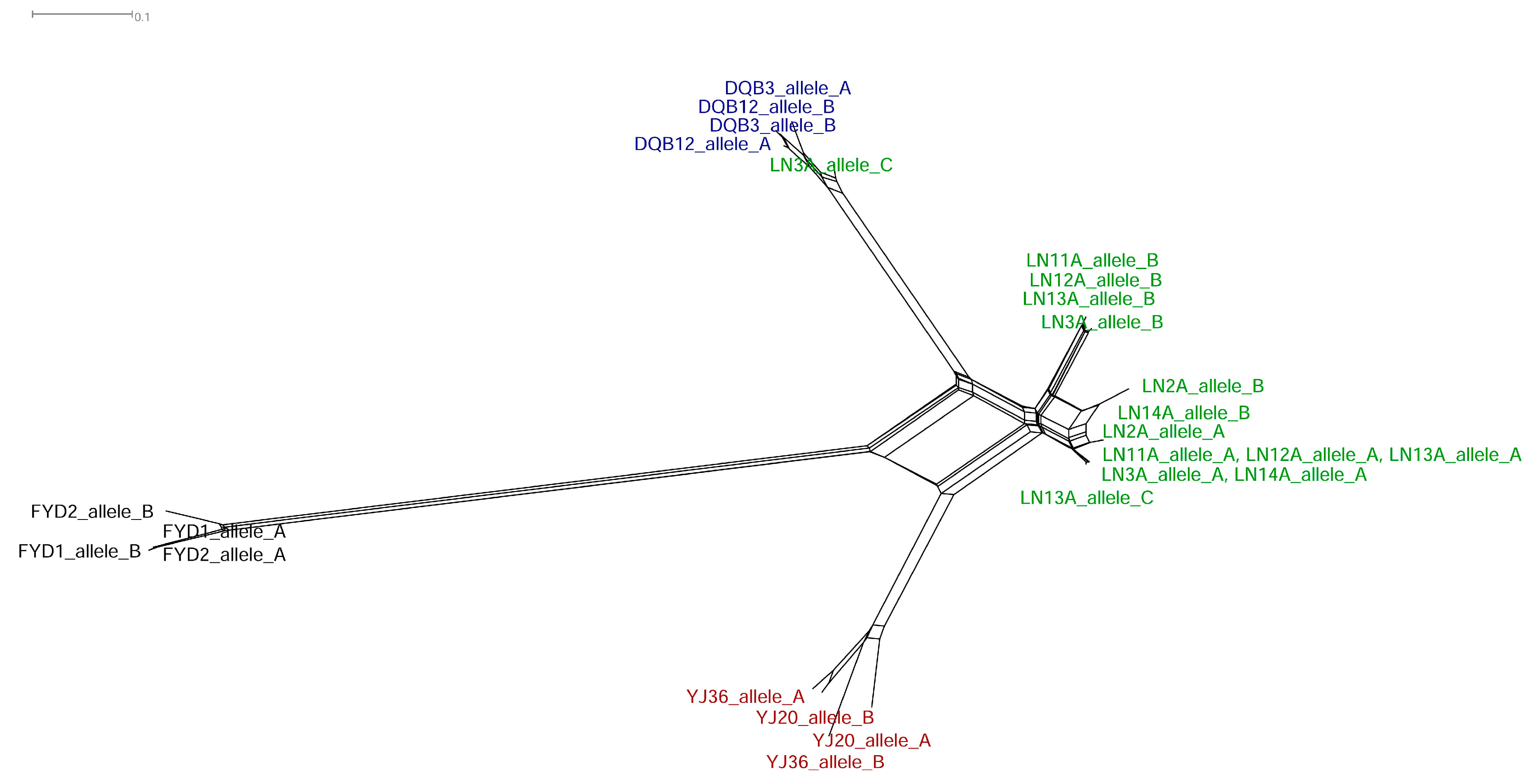
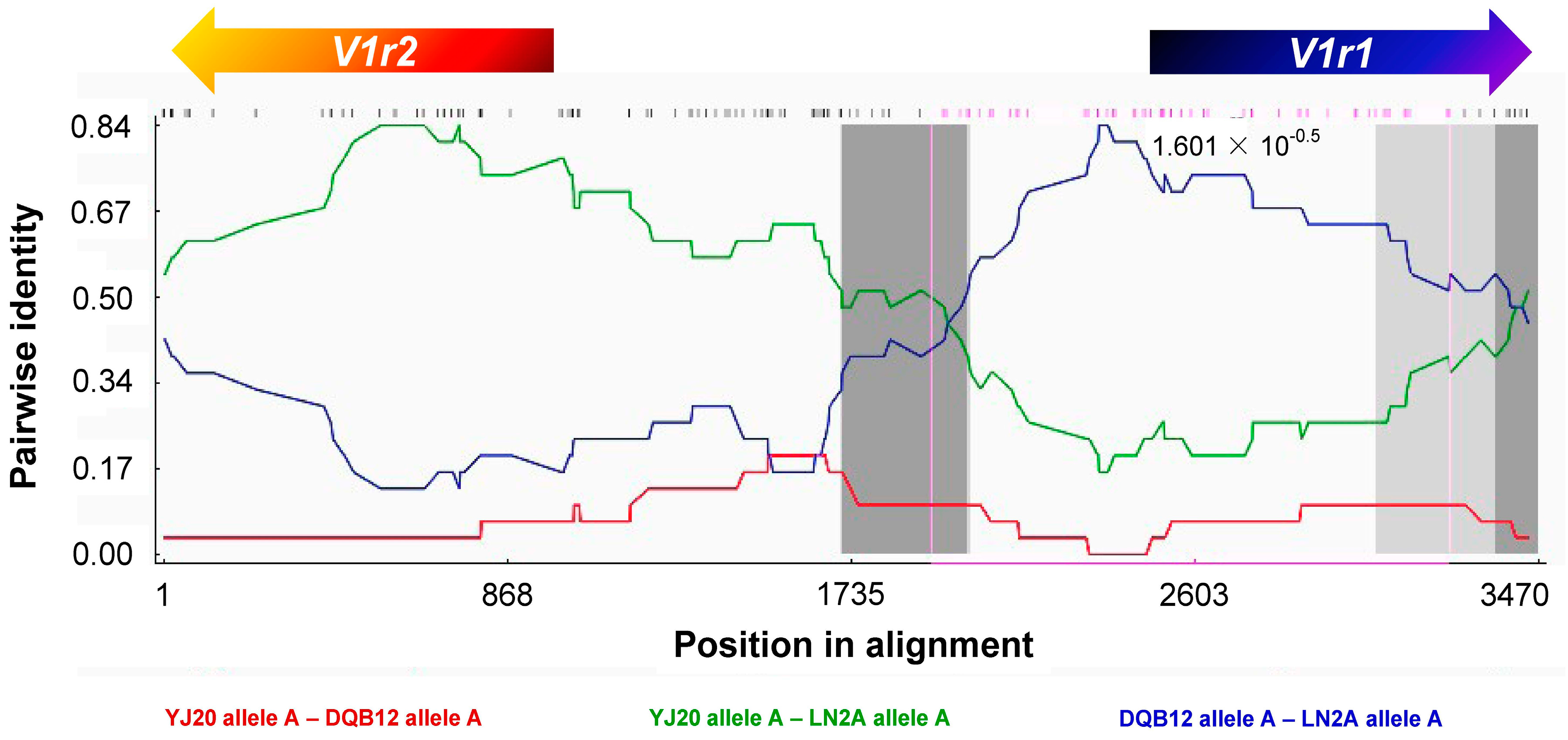
3.3. Recombination between Homoeologous V1r1-V1r2 Clusters in the Tetraploid Genome
3.4. Divergence of V1r1 and V1r2 among the Diploid Parental Species and the Tetraploids
3.5. Heterogeneous Evolution between Nuclear Genes
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bear, D.M.; Lassance, J.M.; Hoekstra, H.E.; Datta, S.R. The Evolving Neural and Genetic Architecture of Vertebrate Olfaction. Curr. Biol. 2016, 26, R1039–R1049. [Google Scholar] [CrossRef] [PubMed]
- Grus, W.E.; Zhang, J.Z. Origin of the Genetic Components of the Vomeronasal System in the Common Ancestor of all Extant Vertebrates. Mol. Biol. Evol. 2009, 26, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, L.R.; Korsching, S.I. A novel olfactory receptor gene family in teleost fish. Genome Res. 2007, 17, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Pfister, P.; Randall, J.; Montoya-Burgos, J.I.; Rodriguez, I. Divergent evolution among teleost V1r receptor genes. PLoS ONE 2007, 2, e379. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.; Frank, O.; Rawel, H.; Ahuja, G.; Potting, C.; Hofmann, T.; Meyerhof, W.; Korsching, S. ORA1, a Zebrafish Olfactory Receptor Ancestral to All Mammalian V1R Genes, Recognizes 4-Hydroxyphenylacetic Acid, a Putative Reproductive Pheromone. J. Biol. Chem. 2014, 289, 19778–19788. [Google Scholar] [CrossRef] [PubMed]
- Zapilko, V.; Korsching, S.I. Tetrapod V1R-like ORA genes in an early-diverging ray-finned fish species: The canonical six ORA gene repertoire of teleost fish resulted from gene loss in a larger ancestral repertoire. BMC Genomics 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, M.; Ota, T.; Hirata, T.; Suzuki, H.; Satta, Y.; Aibara, M.; Mzighani, S.I.; Sturmbauer, C.; Hagino-Yamagishi, K.; Okada, N. Multiple Episodic Evolution Events in V1R Receptor Genes of East-African Cichlids. Genome Biol. Evol. 2014, 6, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; van de Peer, Y. From 2R to 3R: Evidence for a fish-specific genome duplication (FSGD). Bioessays 2005, 27, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Chain, F.J.; Dushoff, J.; Evans, B.J. The odds of duplicate gene persistence after polyploidization. BMC Genomics 2011, 12, 599. [Google Scholar] [CrossRef] [PubMed]
- Roulin, A.; Auer, P.L.; Libault, M.; Schlueter, J.; Farmer, A.; May, G.; Stacey, G.; Doerge, R.W.; Jackson, S.A. The fate of duplicated genes in a polyploid plant genome. Plant J. 2013, 73, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. A study on the classification of the subfamily cobitinae of china. Trans. Chin. Ichthyol. Soc. 1981, 1, 21–32. [Google Scholar]
- Yu, Y.Y.; Abbas, K.; Wang, W.M.; Zhou, X.Y. Geographical distribution of ploidy level variation of loach Misgurnus anguillicaudatus in China. Pak. J. Agric. Sci. 2014, 51, 273–281. [Google Scholar]
- Yi, S.; Zhong, J.; Huang, S.; Wang, S.; Wang, W. Morphological comparison and DNA barcoding of four closely related species in the genera Misgurnus and Paramisgurnus (Cypriniformes: Cobitidae). Biochem. Syst. Ecol. 2017, 70, 50–59. [Google Scholar] [CrossRef]
- Fujimoto, T.; Nishimura, T.; Goto-Kazeto, R.; Kawakami, Y.; Yamaha, E.; Arai, K. Sexual dimorphism of gonadal structure and gene expression in germ cell-deficient loach, a teleost fish. Proc. Natl. Acad. Sci. USA 2010, 107, 17211–17216. [Google Scholar] [CrossRef] [PubMed]
- Pfister, P.; Rodriguez, I. Olfactory expression of a single and highly variable V1r pheromone receptor-like gene in fish species. Proc. Natl. Acad. Sci. USA 2005, 102, 5489–5494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; Mcginnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K. TMBASE—A database of membrane spanning protein segments. Biol. Chem. 1993, 374, 1–3. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. Neighbor-net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrov. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. Evaluation of methods for detecting recombination from DNA sequences: Empirical data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Gojobori, T. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 1999, 16, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.Y.; Frost, S.D.W.; Pond, S.L.K. Detecting Signatures of Selection from DNA Sequences Using Datamonkey. Method Mol. Biol. 2009, 537, 163–183. [Google Scholar]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Kreitman, M.; Aguadé, M. A Test of Neutral Molecular Evolution Based on Nucleotide Data. Genetics 1987, 116, 153–159. [Google Scholar] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Glover, N.M.; Redestig, H.; Dessimoz, C. Homoeologs: What Are They and How Do We Infer Them? Trends Plant. Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Twyford, A.D.; Ennos, R.A. Next-generation hybridization and introgression. Heredity 2012, 108, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Touhara, K.; Vosshall, L.B. Sensing Odorants and Pheromones with Chemosensory Receptors. Annu. Rev. Physiol. 2009, 71, 307–332. [Google Scholar] [CrossRef] [PubMed]
- Malnic, B.; Hirono, J.; Sato, T.; Buck, L.B. Combinatorial receptor codes for odors. Cell. 1999, 96, 713–723. [Google Scholar] [CrossRef]
- Haga-Yamanaka, S.; Ma, L.M.; He, J.; Qiu, Q.; Lavis, L.D.; Looger, L.L.; Yu, C.R. Integrated action of pheromone signals in promoting courtship behavior in male mice. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Katada, S.; Hirokawa, T.; Oka, Y.; Suwa, M.; Touhara, K. Structural basis for a broad but selective ligand spectrum of a mouse olfactory receptor: Mapping the odorant-binding site. J. Neurosci. 2005, 25, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.R.; Yoon, K.; Ko, D.J.; Smith, A.D.; Qiao, M.; Suresh, U.; Burns, S.C.; Penalva, L.O.F. Before It Gets Started: Regulating Translation at the 5’ UTR. Comp. Funct. Genom. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample Number | Sex | Location | Accession Number | |||
|---|---|---|---|---|---|---|---|
| Cyt b | RAG-1 | V1r1 | V1r2 | ||||
| Misgurnus anguillicaudatus (tetraploid race) | LN2A | ♀ | Luoning, Henan | KX189347 | KX189359, KX189360, KX189361 | KX189377, KX189378 | KX189413, KX189414 |
| LN3A | ♀ | Luoning, Henan | KX189348 | KX189362, KX189363, KX189364, KX189365 | KX189379, KX189380, KX189381 | KX189415, KX189416, KX189417 | |
| LN11A | ♂ | Luoning, Henan | KX189349 | KX189366, KX189367, KX189368, KX189369 | KX189392, KX189393 | KX189418, KX189419 | |
| LN12A | ♂ | Luoning, Henan | KX189350 | KX189370, KX189371, KX189372 | KX189394, KX189395 | KX189420, KX189421 | |
| LN13A | ♀ | Luoning, Henan | KX189351 | KX189373, KX189374, KX189375 | KX189396, KX189397, KX189398 | KX189422, KX189423, KX189424 | |
| LN14A | ♂ | Luoning, Henan | KX189352 | KX189376, KX189377, KX189378 | KX189399, KX189400 | KX189425, KX189426 | |
| Misgurnus anguillicaudatus (diploid race) | YJ20 | ♂ | Yueyang, Hunan | KX189353 | KX189379, KX189380 | KX189401, KX189402 | KX189427, KX189428 |
| YJ36 | ♀ | Yueyang, Hunan | KX189354 | KX189381 | KX189403, KX189404 | KX189429, KX189430 | |
| Misgurnus bipartitus (diploid) | DQB3 | ♀ | Daqing, Helongjiang | KX189355 | KX189382, KX189383 | KX189405, KX189406 | KX189431, KX189432 |
| DQB12 | ♂ | Daqing, Heilongjiang | KX189356 | KX189384 | KX189407, KX189408 | KX189433, KX189434 | |
| Paramisgurnus dabryanus (diploid) | FYD1 | ♀ | Fuyuan, Heilongjiang | KX189357 | KX189385 | KX189409, KX189410 | KX189435, KX189436 |
| FYD2 | ♂ | Fuyuan, Heilongjiang | KX189358 | KX189386 | KX189411, KX189412 | KX189437, KX189438 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, L.; Wang, W. Homoeologous Recombination of the V1r1-V1r2 Gene Cluster of Pheromone Receptors in an Allotetraploid Lineage of Teleosts. Genes 2017, 8, 334. https://doi.org/10.3390/genes8110334
Zhong L, Wang W. Homoeologous Recombination of the V1r1-V1r2 Gene Cluster of Pheromone Receptors in an Allotetraploid Lineage of Teleosts. Genes. 2017; 8(11):334. https://doi.org/10.3390/genes8110334
Chicago/Turabian StyleZhong, Lei, and Weimin Wang. 2017. "Homoeologous Recombination of the V1r1-V1r2 Gene Cluster of Pheromone Receptors in an Allotetraploid Lineage of Teleosts" Genes 8, no. 11: 334. https://doi.org/10.3390/genes8110334
APA StyleZhong, L., & Wang, W. (2017). Homoeologous Recombination of the V1r1-V1r2 Gene Cluster of Pheromone Receptors in an Allotetraploid Lineage of Teleosts. Genes, 8(11), 334. https://doi.org/10.3390/genes8110334