Human Organ Tissue Identification by Targeted RNA Deep Sequencing to Aid the Investigation of Traumatic Injury


Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Body Fluid Stains
2.2. RNA Isolation
2.3. DNase I Digestion
2.4. RNA Quantification
2.5. TruSeq® Targeted RNA Library Preparation
2.6. TruSeq® Targeted RNA Library Quantification
2.7. MiSeq® Sequencing
2.8. Data Analysis
3. Results
3.1. Assay Development
3.1.1. Candidate Selection
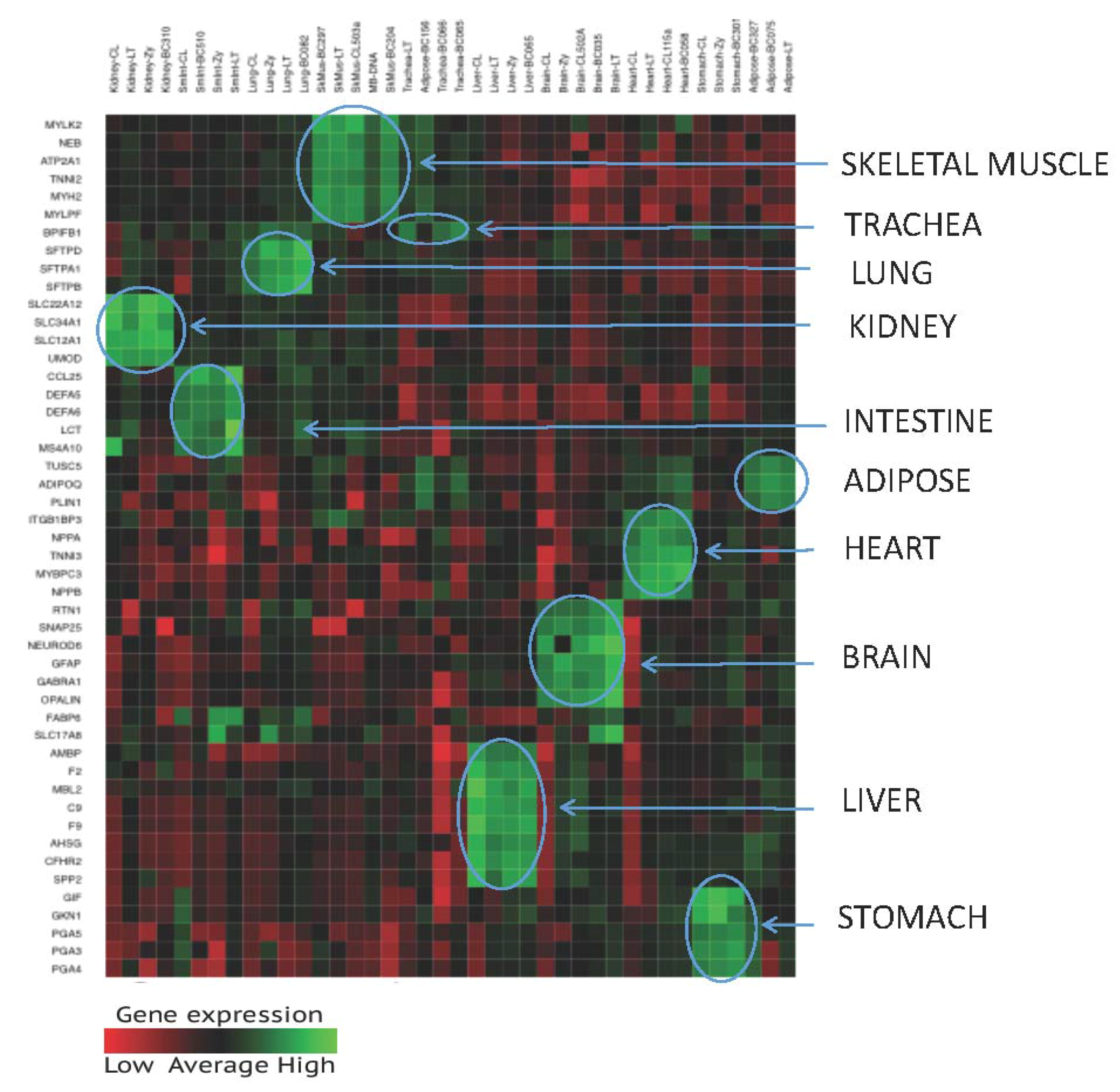
3.1.2. Specificity of the 46—Plex Targeted RNA Sequencing Assay
3.1.3. Tissue Inference
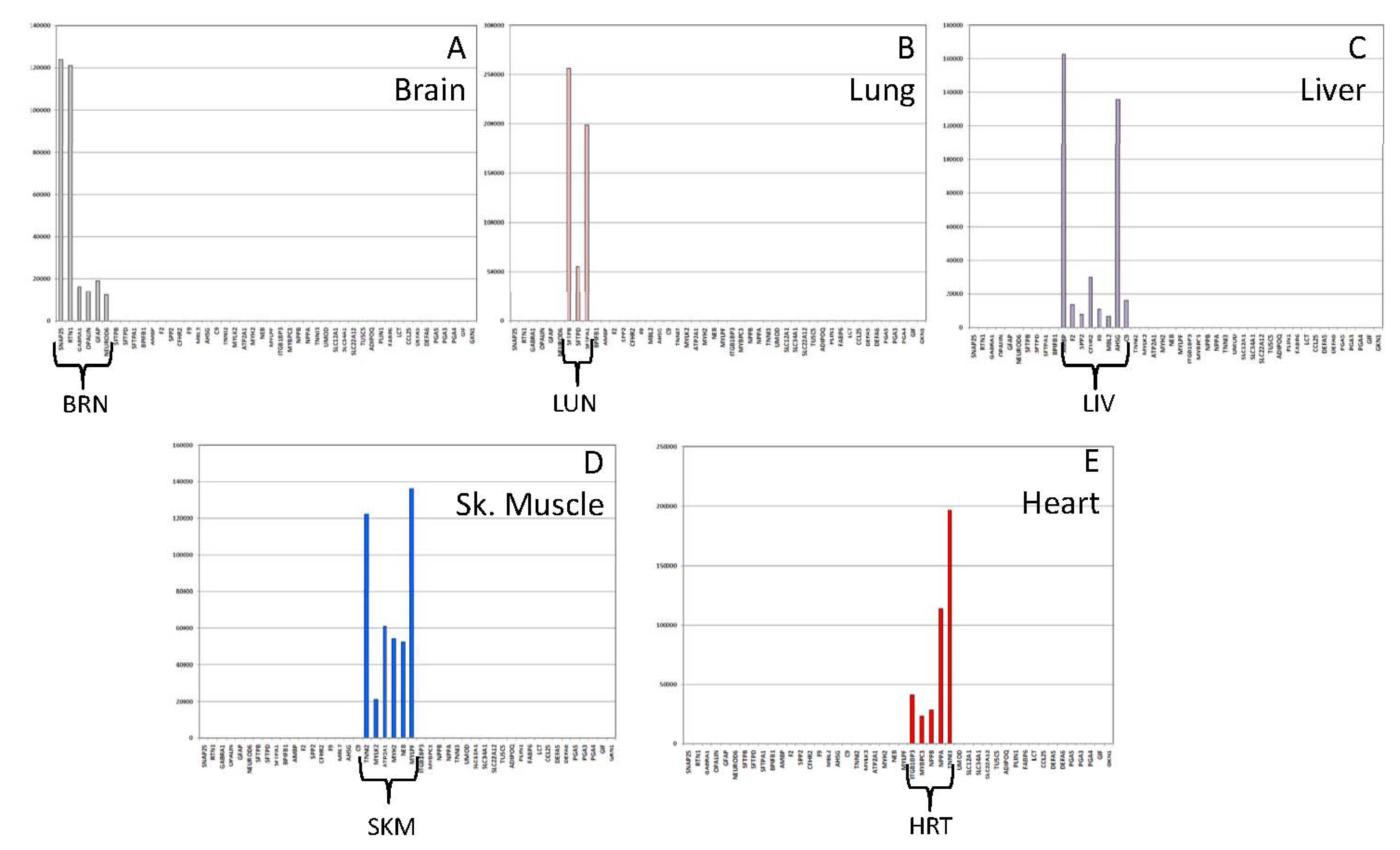
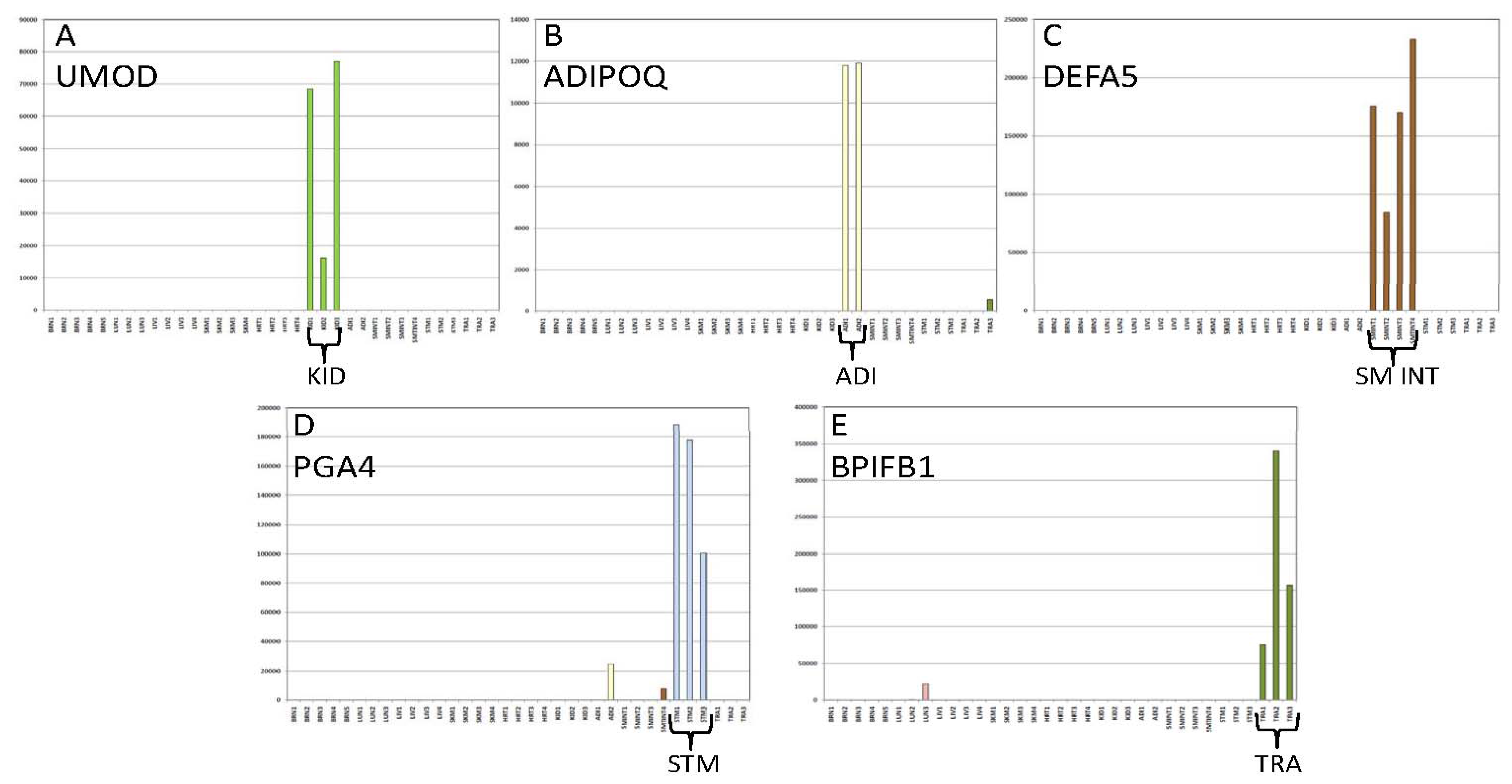
3.2. Performance Testing
3.2.1. Biomarker Sensitivity of Detection
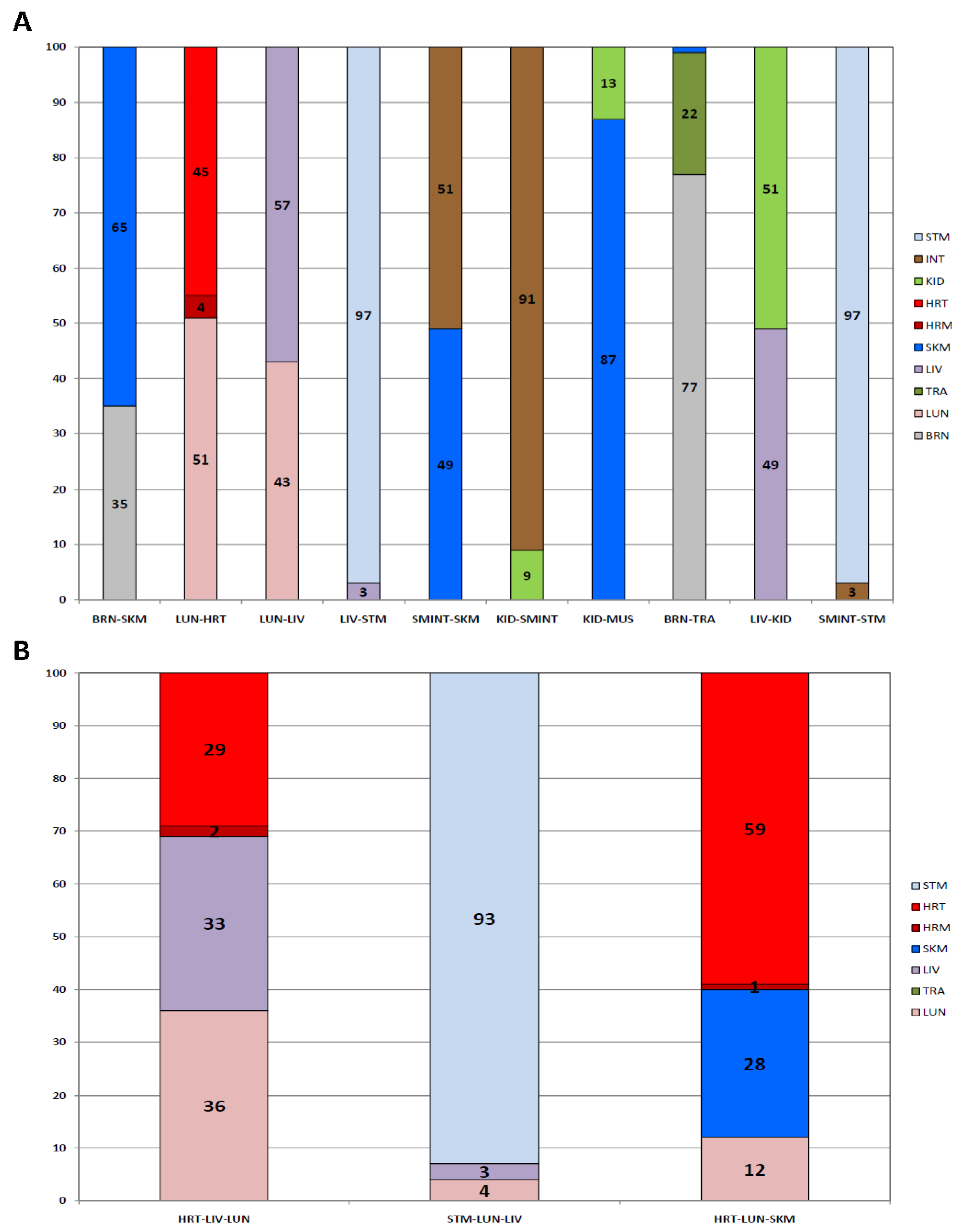
3.2.2. Mixtures
3.2.3. Repeatability
3.2.4. Specificity
3.2.5. DNA and Amplification Blanks
3.3. Blind Study
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DiMaio, V.J.M. Gunshot Wounds: Practical Aspects of Firearms, Ballistics and Forensic Techniques; CRC Press: Boca Raton, FL, USA, 2015; pp. 1–377. ISBN 978-1498725694. [Google Scholar]
- Prahlow, J.A.; Byard, R.W. Atlas of Forensic Pathology; Springer: New York, NY, USA, 2012; pp. 1–85. ISBN 978-1-61779-057-7. [Google Scholar]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 1994; pp. 1–1352. ISBN 978-0815316206. [Google Scholar]
- Sauer, E.; Extra, A.; Cachee, P.; Courts, C. Identification of organ tissue types and skin from forensic samples by microRNA expression analysis. Forensic Sci. Int. Genet. 2017, 28, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Patzelt, D. Evaluation of mRNA markers for the identification of menstrual blood. J. Forensic Sci. 2002, 47, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Patzelt, D. Protamine mRNA as molecular marker for spermatozoa in semen stains. Int. J. Legal Med. 2003, 117, 175–179. [Google Scholar] [PubMed]
- Fleming, R.I.; Harbison, S. The development of a mRNA multiplex RT-PCR assay for the definitive identification of body fluids. Forensic Sci. Int. Genet. 2010, 4, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Frumkin, D.; Wasserstrom, A.; Budowle, B.; Davidson, A. DNA methylation-based forensic tissue identification. Forensic Sci. Int. Genet. 2011, 5, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Klesser, B.; Maake, C.; Bar, W.; Kratzer, A. mRNA profiling for body fluid identification by reverse transcription endpoint PCR and realtime PCR. Forensic Sci. Int. Genet. 2009, 3, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Hanson, E.; Ballantyne, J. Capillary electrophoresis of a multiplex reverse transcription-polymerase chain reaction to target messenger RNA markers for body fluid identification. Methods Mol. Biol. 2012, 830, 169–183. [Google Scholar] [PubMed]
- Hanson, E.; Lubenow, H.; Ballantyne, J. Identification of forensically relevant body fluids using a panel of differentially expressed microRNAs. Forensic Sci. Int. Genet. 2009, 2, 503–504. [Google Scholar] [CrossRef]
- Hanson, E.; Haas, C.; Jucker, R.; Ballantyne, J. Identification of skin in touch/contact forensic samples by messenger RNA profiling. Forensic Sci. Int. Genet. Supp. Ser. 2011, 3, e305–e306. [Google Scholar] [CrossRef]
- Hanson, E.; Haas, C.; Jucker, R.; Ballantyne, J. Specific and sensitive mRNA biomarkers for the identification of skin in 'touch DNA' evidence. Forensic Sci. Int. Genet. 2012, 6, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Ingold, S.; Haas, C.; Ballantyne, J. Targeted multiplexed next generation RNA sequencing assay for tissue source determination of forensic samples. Forensic Sci. Int. Genet. Supp. Ser. 2015, 5, e441–e443. [Google Scholar] [CrossRef]
- Hanson, E.K.; Ballantyne, J. RNA Profiling for the Identification of the Tissue Origin of Dried Stains in Forensic Biology. Forensic Sci. Rev. 2010, 22, 145–157. [Google Scholar] [PubMed]
- Hanson, E.K.; Ballantyne, J. Rapid and inexpensive body fluid identification by RNA profiling-based multiplex High Resolution Melt (HRM) analysis. F1000Research 2013, 2, 281. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.K.; Mirza, M.; Rekab, K.; Ballantyne, J. The identification of menstrual blood in forensic samples by logistic regression modeling of miRNA expression. Electrophoresis 2014, 35, 3087–3095. [Google Scholar] [CrossRef] [PubMed]
- Juusola, J.; Ballantyne, J. Messenger RNA profiling: A prototype method to supplant conventional methods for body fluid identification. Forensic Sci. Int. 2003, 135, 85–96. [Google Scholar] [CrossRef]
- Juusola, J.; Ballantyne, J. Multiplex mRNA profiling for the identification of body fluids. Forensic Sci. Int. 2005, 152, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Juusola, J.; Ballantyne, J. mRNA profiling for body fluid identification by multiplex quantitative RT-PCR. J. Forensic Sci. 2007, 52, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Park, M.J.; Choi, A.; An, J.H.; Yang, W.I.; Shin, K.J. Potential forensic application of DNA methylation profiling to body fluid identification. Int. J. Legal Med. 2012, 126, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Legg, K.M.; Powell, R.; Reisdorph, N.; Reisdorph, R.; Danielson, P.B. Discovery of highly specific protein markers for the identification of biological stains. Electrophoresis 2014, 35, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, A.; de Pagter, M.; Ramdayal, G.; Visser, M.; Zubakov, D.; Kayser, M.; Sijen, T. A multiplex (m)RNA-profiling system for the forensic identification of body fluids and contact traces. Forensic Sci. Int. Genet. 2012, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, A.; van den Berge, M.; Oostra, R.J.; Cleypool, C.; Bruggink, A.; Kloosterman, A.; Sijen, T. Development of a mRNA profiling multiplex for the inference of organ tissues. Int. J. Legal Med. 2013, 127, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, A.; Maaskant, P.; Sijen, T. Implementation of RNA profiling in forensic casework. Forensic Sci. Int. Genet. 2013, 7, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Madi, T.; Balamurugan, K.; Bombardi, R.; Duncan, G.; McCord, B. The determination of tissue-specific DNA methylation patterns in forensic biofluids using bisulfite modification and pyrosequencing. Electrophoresis 2012, 33, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Harper, K.A.; Craig, R.L.; Onorato, A.J.; Robertson, J.M.; Donfack, J. Evaluation of mRNA marker specificity for the identification of five human body fluids by capillary electrophoresis. Forensic Sci. Int. Genet. 2012, 6, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Roeder, A.D.; Haas, C. mRNA profiling using a minimum of five mRNA markers per body fluid and a novel scoring method for body fluid identification. Int. J. Legal Med. 2013, 127, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Sijen, T. Molecular approaches for forensic cell type identification: On mRNA, miRNA, DNA methylation and microbial markers. Forensic Sci. Int. Genet. 2015, 18, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, M.; Carracedo, A.; Gomes, I.; Graham, E.A.; Haas, C.; Hjort, B.; Hoff-Olsen, P.; Maronas, O.; Mevag, B.; Morling, N.; et al. A collaborative European exercise on mRNA-based body fluid/skin typing and interpretation of DNA and RNA results. Forensic Sci. Int. Genet. 2014, 10, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, M.; Ozcanhan, G.; Zijlstra, S.; Lindenbergh, A.; Sijen, T. Prevalence of human cell material: DNA and RNA profiling of public and private objects and after activity scenarios. Forensic Sci. Int. Genet. 2015, 21, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, M.; Bhoelai, B.; Harteveld, J.; Matai, A.; Sijen, T. Advancing forensic RNA typing: On non-target secretions, a nasal mucosa marker, a differential co-extraction protocol and the sensitivity of DNA and RNA profiling. Forensic Sci. Int. Genet. 2016, 20, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Van Steendam, K.; De Ceuleneer, M.; Dhaenens, M.; Van Hoofstat, D.; Deforce, D. Mass spectrometry-based proteomics as a tool to identify biological matrices in forensic science. Int. J. Legal Med. 2013, 127, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, B.; Deng, H.; Prinz, M.; Siegel, D. Body fluid identification by mass spectrometry. Int. J. Legal Med. 2013, 127, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Zubakov, D.; Hanekamp, E.; Kokshoorn, M.; van Ijcken, W.; Kayser, M. Stable RNA markers for identification of blood and saliva stains revealed from whole genome expression analysis of time-wise degraded samples. Int. J. Legal Med. 2008, 122, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Zubakov, D.; Kokshoorn, M.; Kloosterman, A.; Kayser, M. New markers for old stains: Stable mRNA markers for blood and saliva identification from up to 16-year-old stains. Int. J. Legal Med. 2009, 123, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Zubakov, D.; Boersma, A.W.; Choi, Y.; van Kuijk, P.F.; Wiemer, E.A.; Kayser, M. MicroRNA markers for forensic body fluid identification obtained from microarray screening and quantitative RT-PCR confirmation. Int. J. Legal Med. 2010, 124, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Dammeier, S.; Nahnsen, S.; Veit, J.; Wehner, F.; Ueffing, M.; Kohlbacher, O. Mass-spectrometry-based proteomics reveals organ-specific expression patterns to be used as forensic evidence. J. Proteome Res. 2016, 15, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, J.; Lindenbergh, A.; Sijen, T. RNA cell typing and DNA profiling of mixed samples: Can cell types and donors be associated? Sci. Justice 2013, 53, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Murty, M.N.; Flynn, P.J. Data clustering: A review. ACM Comput. Surv. 1999, 31, 264–323. [Google Scholar] [CrossRef]
- Hanson, E.K.; Ingold, S.; Haas, C.; Ballantyne, J. Messenger RNA Biomarker Signatures for Forensic Body Fluid Identification Revealed by Targeted RNA Sequencing. Forensic Sci. Int. Genet. 2017. under review. [Google Scholar]
- Dorum, G.; Ingold, S.; Hanson, E.; Ballantyne, J.; Snipen, L.; Haas, C. Predicting the Origin of Stains from Next Generation Sequencing mRNA Data. Forensic Sci. Int. Genet. 2017. under review. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Gene Name | Chromosome | Transcript ID | Illumina Assay ID |
|---|---|---|---|---|
| Brain | SNAP25 | 20 | NM_130811 | 6650651 |
| RTN1 | 14 | NM_021136 | 6597471 | |
| GABRA1 | 5 | NM_001127643 | 6769405 | |
| OPALIN | 10 | NM_001040103 | 6690750 | |
| GFAP | 17 | NM_002055 | 6760207 | |
| NEUROD6 | 7 | NM_022728 | 6608149 | |
| Lung | SFTPB | 2 | NM_198843 | 6822231 |
| SFTPD | 10 | NM_003019 | 6635044 | |
| SFTPA1 | 10 | NM_005411 | 6736962 | |
| Trachea | BPIFB1 | 20 | NM_033197 | 6804173 |
| Liver | AMBP | 9 | NM_001633 | 6846165 |
| F2 | 11 | NM_000506 | 6834705 | |
| SPP2 | 2 | NM_006944 | 6646626 | |
| CFHR2 | 1 | NM_005666 | 6824671 | |
| F9 | X | NM_000133 | 6813125 | |
| MBL2 | 10 | NM_000242 | 6748563 | |
| AHSG | 3 | NM_001622 | 6842654 | |
| C9 | 5 | NM_001737 | 6711440 | |
| Skeletal | TNNI2 | 11 | NM_003282 | 6650981 |
| Muscle | MYLK2 | 20 | NM_033118 | 6800284 |
| ATP2A1 | 16 | NM_004320 | 6782675 | |
| MYH2 | 17 | NM_017534 | 6700111 | |
| NEB | 2 | NM_001164508 | 6690232 | |
| MYLPF | 16 | NM_013292 | 6688633 | |
| Heart Muscle | ITGB1BP3 | 19 | NM_170678 | 6650498 |
| Heart | MYBPC3 | 11 | NM_000256 | 6685046 |
| NPPB | 1 | NM_002521 | 6847931 | |
| NPPA | 1 | NM_006172 | 6634864 | |
| TNNI3 | 19 | NM_000363 | 6715646 | |
| Kidney | UMOD | 16 | NM_003361 | 6842087 |
| SLC12A1 | 15 | NM_001184832 | 6692344 | |
| SLC34A1 | 5 | NM_003052 | 6850242 | |
| SLC22A12 | 11 | NM_153378 | 6678522 | |
| Adipose | TUSC5 | 17 | NM_172367 | 6779317 |
| ADIPOQ | 3 | NM_001177800 | 6795292 | |
| PLIN1 | 15 | NM_002666 | 6654705 | |
| Intestine | FABP6 | 5 | NM_001130958 | 6641583 |
| LCT | 2 | NM_002299 | 6648509 | |
| CCL25 | 19 | NM_005624 | 6726865 | |
| DEFA5 | 8 | NM_021010 | 6669611 | |
| DEFA6 | 8 | NM_001926 | 6625127 | |
| Stomach | PGA5 | 11 | NM_014224 | 6775995 |
| PGA3 | 11 | NM_001079807 | 6973516 | |
| PGA4 | 11 | NM_001079808 | 6983051 | |
| GIF | 11 | NM_005142 | 6675517 | |
| GKN1 | 2 | NM_019617 | 6798784 |
| Brain | Lung | Trachea | Liver | Sk.Mus | Heart | Kidney | Adipose | Sm.Int | Stomach | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | 5 | 3 | 3 | 4 | 4 | 4 | 3 | 2 | 4 | 3 | |
| Avg Total | 342,617 | 353,031 | 210,086 | 331,286 | 415,965 | 383,774 | 118,472 | 104,731 | 324,757 | 515,896 | |
| BRN | SNAP25 | 167,707 | |||||||||
| RTN1 | 83,980 | * | 1927 | 2045 | |||||||
| GABRA1 | 14,584 | ||||||||||
| OPALIN | 9698 | ||||||||||
| GFAP | 53,931 | ||||||||||
| NEUROD6 | 9872(4) | ||||||||||
| LUN | SFTPB | 145,365 | * | ||||||||
| SFTPD | 56,487 | ||||||||||
| STFPA1 | 142,360 | * | |||||||||
| TRA | BPIFB1 | 11,338(2) | 190,738 | ||||||||
| LIV | AMBP | * | 165,375 | * | |||||||
| F2 | 13,915 | ||||||||||
| SPP2 | 6787(3) | ||||||||||
| CFHR2 | 21,586 | ||||||||||
| F9 | 9090 | ||||||||||
| MBL2 | 4649(3) | * | |||||||||
| AHSG | * | 93,723 | |||||||||
| C9 | 19,021 | ||||||||||
| SKM | TNNI2 | * | 106,756 | * | |||||||
| MYLK2 | 19,082 | ||||||||||
| ATP2A1 | 53,200 | * | |||||||||
| MYH2 | 1586(2) | 43,511 | * | ||||||||
| NEB | 2491(2) | 67,115 | * | ||||||||
| MYLPF | 1583(2) | 125,702 | * | ||||||||
| HRT | ITGB1BP3 | * | 20,005 | ||||||||
| MYBPC3 | 17,803 | ||||||||||
| NPPB | 14,539 | ||||||||||
| NPPA | * | 162,719 | |||||||||
| TNNI3 | * | 168,146 | |||||||||
| KID | UMOD | 53,914 | |||||||||
| SLC12A1 | 39,341 | ||||||||||
| SLC34A1 | 16,085 | ||||||||||
| SLC22A12 | 9133 | ||||||||||
| ADI | TUSC5 | 6842 | |||||||||
| ADIPOQ | 11,854 | ||||||||||
| PLIN1 | 2533 | * | 21,176 | ||||||||
| INT | FABP6 | 36,487(3) | |||||||||
| LCT | * | ||||||||||
| CCL25 | 4333(3) | ||||||||||
| DEFA5 | 165,872 | ||||||||||
| DEFA6 | 114,731 | ||||||||||
| STM | PGA5 | * | * | 23,954 | |||||||
| PGA3 | * | * | 103,475 | ||||||||
| PGA4 | * | * | 155,582 | ||||||||
| GIF | 7311(2) | ||||||||||
| GKN1 | 18,557(2) |
| Brain | Lung | Trachea | Liver | Sk.Mus | Heart | Kidney | Adipose | Sm.Int | Stomach | |
|---|---|---|---|---|---|---|---|---|---|---|
| Biomarkers | N = 5 | N = 3 | N = 3 | N = 4 | N = 4 | N = 4 | N = 3 | N = 2 | N = 4 | N = 3 |
| BRN | 99(96-99) | 0(0-1) | 1 | 0 | 0 | 0 | 0 | 2(1-3) | 0 | 0 |
| LUN | 0 | 98(94-100) | 1(0-2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TRA | 0 | 2(1-5) | 89(79-98) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| LIV | 1(0-4) | 0 | 0 | 100 | 0 | 0 | 0 | 5(5-10) | 0 | 0 |
| SKM | 0 | 0 | 4(0-8) | 0 | 100(99-100) | 0 | 0 | 19(0-38) | 0 | 0 |
| HRM | 0 | 0 | 4(0-13) | 0 | 0 | 5(2-10) | 0 | 0 | 0 | 0 |
| HRT | 0 | 0 | 0 | 0 | 0 | 95(90-98) | 0 | 0 | 0 | 0 |
| KID | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 |
| ADI | 0 | 0 | 1 | 0 | 0 | 0(0-1) | 0 | 42(25-59) | 0 | 0 |
| INT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 98(92-100) | 0 |
| STM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 32(0-63) | 2(0-8) | 100 |
| Tissue | Input (ng) | Total Reads | % Cont | SNAP25 | RTN1 | GABRA1 | OPALIN | GFAP | NEUROD6 | ||
| Brain | 25 | 253,551 | 99 | 120,086 | 71,499 | 8192 | 8698 | 36,150 | 6901 | ||
| 10 | 68,930 | 100 | 31,986 | 19,041 | 3036 | 2929 | 11,055 | 883 | |||
| 5 | 12,021 | 100 | 7256 | 3315 | 1450 | ||||||
| Tissue | Input (ng) | Total Reads | % Cont | SFTPB | SFTPD | SFTPA1 | |||||
| Lung | 25 | 641,600 | 100 | 232,508 | 31,220 | 377,872 | |||||
| 10 | 103,030 | 100 | 43,874 | 6451 | 52,705 | ||||||
| 5 | 15,448 | 100 | 7612 | 808 | 7028 | ||||||
| Tissue | Input (ng) | Total Reads | % Cont | BPIFB1 | |||||||
| Trachea | 25 | 75,524 | 92 | 69,521 | |||||||
| 10 | 63,490 | 99 | 62,901 | ||||||||
| 5 | Below MTR | -- | |||||||||
| Tissue | Input (ng) | Total Reads | % Cont | AMBP | F2 | SPP2 | CFHR2 | F9 | MBL2 | AHSG | C9 |
| Liver | 25 | 169,000 | 100 | 90.462 | 7565 | 3739 | 11,117 | 4328 | 1541 | 43,900 | 6348 |
| 10 | 47,366 | 100 | 24,898 | 2598 | 695 | 3197 | 870 | 696 | 11,850 | 2562 | |
| 5 | 26,444 | 100 | 15,739 | 539 | -- | 1807 | 592 | -- | 6678 | 1089 | |
| Tissue | Input (ng) | Total Reads | % Cont | TNNI2 | MYLK2 | ATP2A1 | MYH2 | NEB | MYLPF | ||
| Sk. Mus | 25 | 234,740 | 100 | 50,132 | 9910 | 27,752 | 25,867 | 27,058 | 94,021 | ||
| 10 | 7053 | 100 | 706 | -- | 942 | 1031 | 798 | 3576 | |||
| 5 | 20,865 | 100 | 3615 | 768 | 2061 | 2276 | 2190 | 9955 | |||
| Tissue | Input (ng) | Total Reads | % Cont | ITGB1BP3 | MYBPC3 | NPPB | NPPA | TNNI3 | |||
| Heart | 25 | 550,182 | 100 | 46,661 | 30,405 | 29,711 | 149,253 | 294,152 | |||
| 10 | 33,336 | 100 | 2427 | 2115 | 1790 | 8145 | 18,859 | ||||
| 5 | 17,489 | 100 | 765 | 832 | 893 | 5445 | 9554 | ||||
| Tissue | Input (ng) | Total Reads | % Cont | UMOD | SLC12A1 | SLC34A1 | SLC22A12 | ||||
| Kidney | 25 | 30,360 | 100 | 9124 | 9356 | 7107 | 4773 | ||||
| 10 | 14,360 | 100 | 4598 | 4556 | 3269 | 1937 | |||||
| 5 | Below MTR | -- | |||||||||
| Tissue | Input (ng) | Total Reads | % Cont | FABP6 | LCT | CCL25 | DEFA5 | DEFA6 | |||
| Sm. Int | 25 | 1,227,602 | 100 | 189,948 | -- | -- | 690,364 | 347,290 | |||
| 10 | 557,746 | 100 | 74,829 | -- | -- | 314,865 | 167,268 | ||||
| 5 | 398,704 | 100 | 55,869 | -- | -- | 223,287 | 119,549 | ||||
| Tissue | Input (ng) | Total Reads | % Cont | PGA5 | PGA3 | PGA4 | GIF | GKN1 | |||
| Stomach | 25 | 486,450 | 100 | 227,634 | 49,505 | 178,348 | 8322 | 22,641 | |||
| 10 | 147,139 | 100 | 73,155 | 16,109 | 46,184 | 2379 | 9312 | ||||
| 5 | 121,928 | 100 | 66,997 | 8539 | 36,550 | 2241 | 7601 |
| Unk 1 | Unk 2 | Unk 3 | Unk 4 | Unk 5 | Unk 6 | |
|---|---|---|---|---|---|---|
| % Contr. | ||||||
| BRN | 0 | 100 | - | 2 | 0 | 0 |
| LUN | 0 | 0 | - | 0 | 0 | 0 |
| TRA | 0 | 0 | - | 0 | 0 | 0 |
| LIV | 0 | 0 | - | 0 | 0 | 100 |
| SKM | 0 | 0 | - | 11 | 100 | 0 |
| HRM | 0 | 0 | - | 0 | 0 | 0 |
| HRT | 0 | 0 | - | 0 | 0 | 0 |
| KID | 0 | 0 | - | 0 | 0 | 0 |
| ADI | 0 | 0 | - | 86 | 0 | 0 |
| INT | 0 | 0 | - | 0 | 0 | 0 |
| STM | 100 | 0 | - | 1 | 0 | 0 |
| Analyst Conclusion | Stomach | Brain | No tissue detected | Adipose | Skeletal Muscle | Liver |
| Actual | Stomach | Brain (poly A) | Blank (water) | Adipose | Skeletal Muscle | Liver (fetal) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanson, E.; Ballantyne, J. Human Organ Tissue Identification by Targeted RNA Deep Sequencing to Aid the Investigation of Traumatic Injury. Genes 2017, 8, 319. https://doi.org/10.3390/genes8110319
Hanson E, Ballantyne J. Human Organ Tissue Identification by Targeted RNA Deep Sequencing to Aid the Investigation of Traumatic Injury. Genes. 2017; 8(11):319. https://doi.org/10.3390/genes8110319
Chicago/Turabian StyleHanson, Erin, and Jack Ballantyne. 2017. "Human Organ Tissue Identification by Targeted RNA Deep Sequencing to Aid the Investigation of Traumatic Injury" Genes 8, no. 11: 319. https://doi.org/10.3390/genes8110319
APA StyleHanson, E., & Ballantyne, J. (2017). Human Organ Tissue Identification by Targeted RNA Deep Sequencing to Aid the Investigation of Traumatic Injury. Genes, 8(11), 319. https://doi.org/10.3390/genes8110319