
Postnatal Gene Therapy Improves Spatial Learning Despite the Presence of Neuronal Ectopia in a Model of Neuronal Migration Disorder

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Viral Vectors and Injection
2.3. Antibodies and Wisteria Floribunda Lectin
2.4. Western Blot Analysis and Ligand Overlay Experiments
2.5. Fluorescence Staining
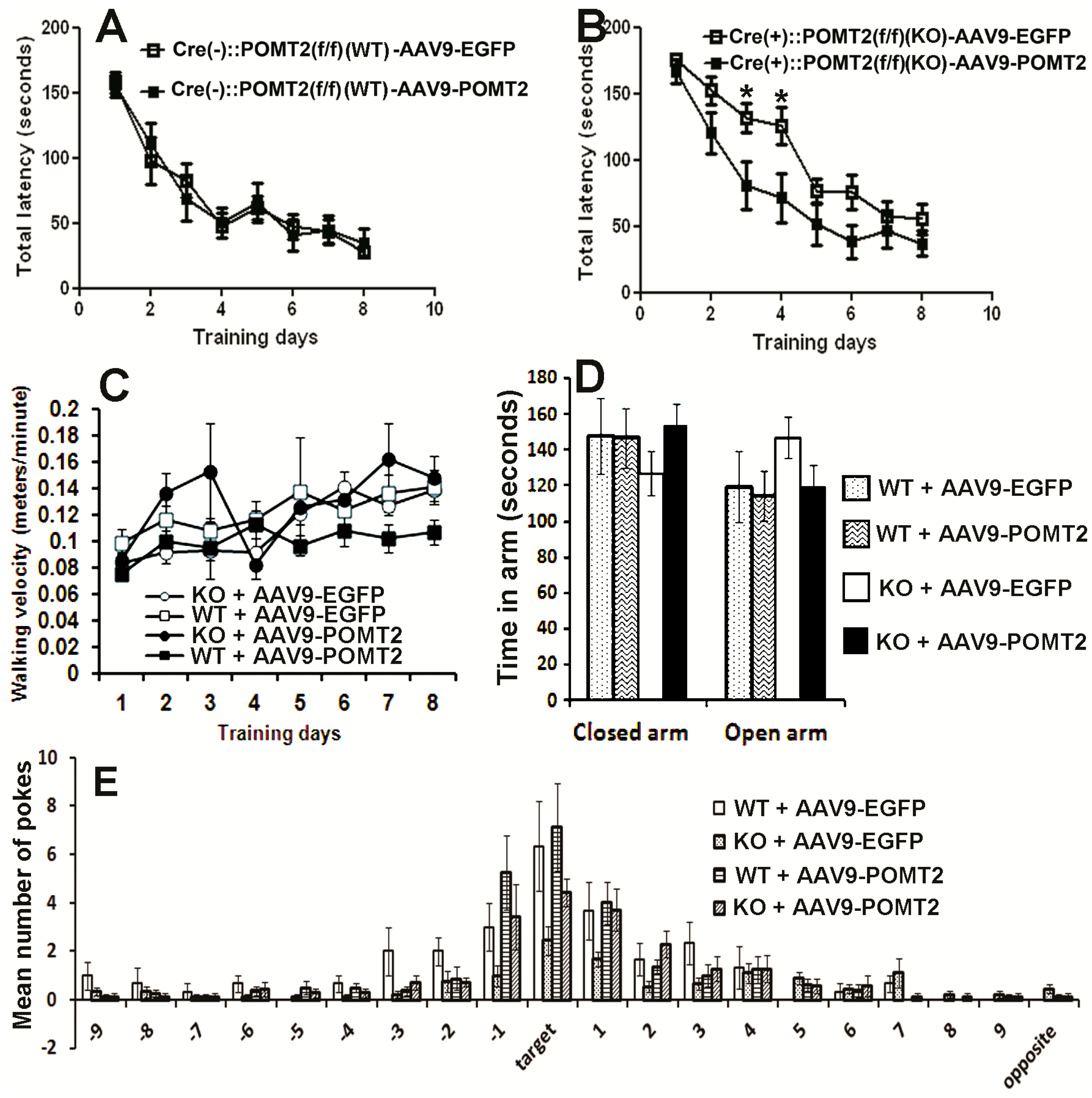
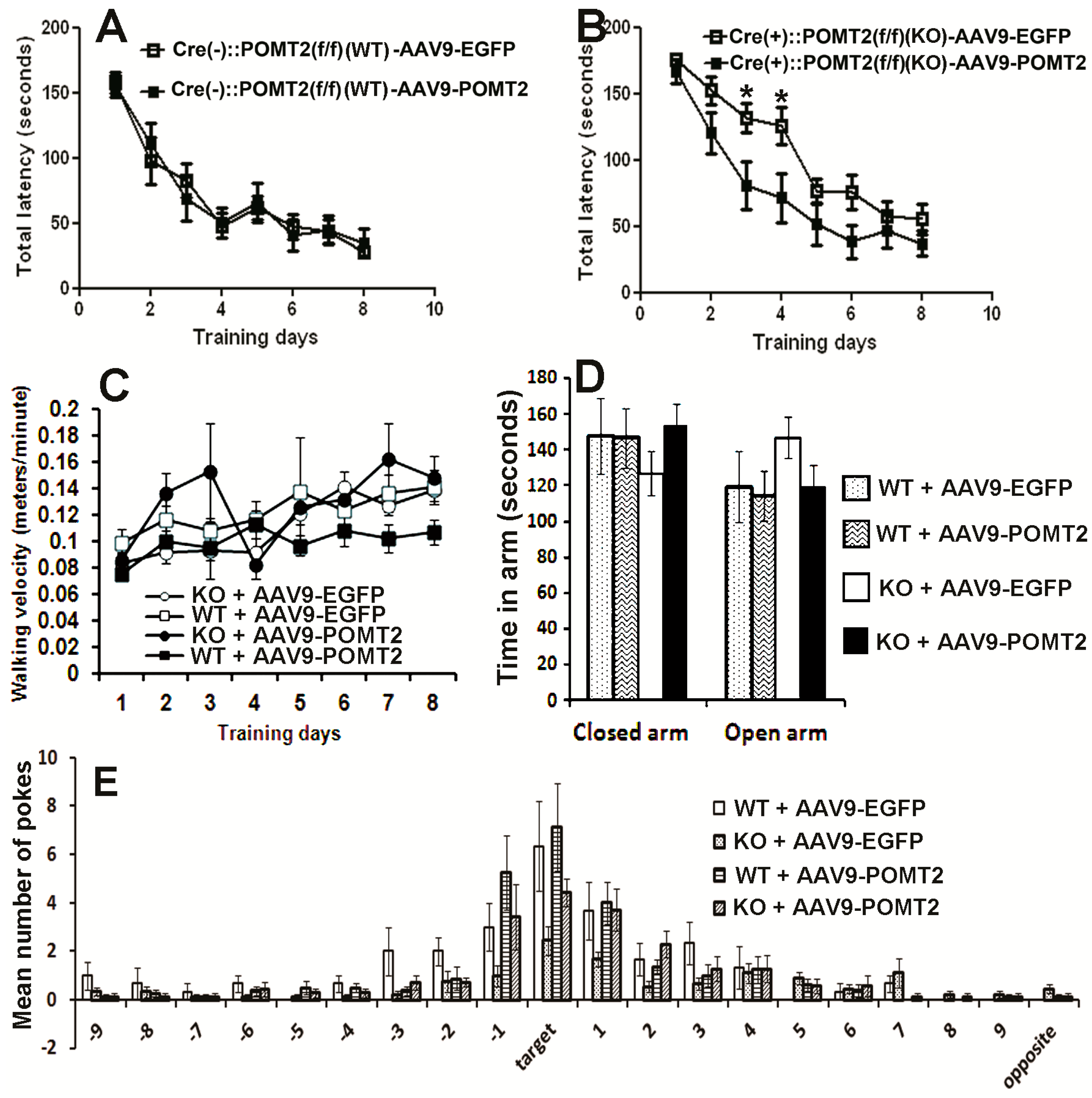
2.6. Behavioral Test
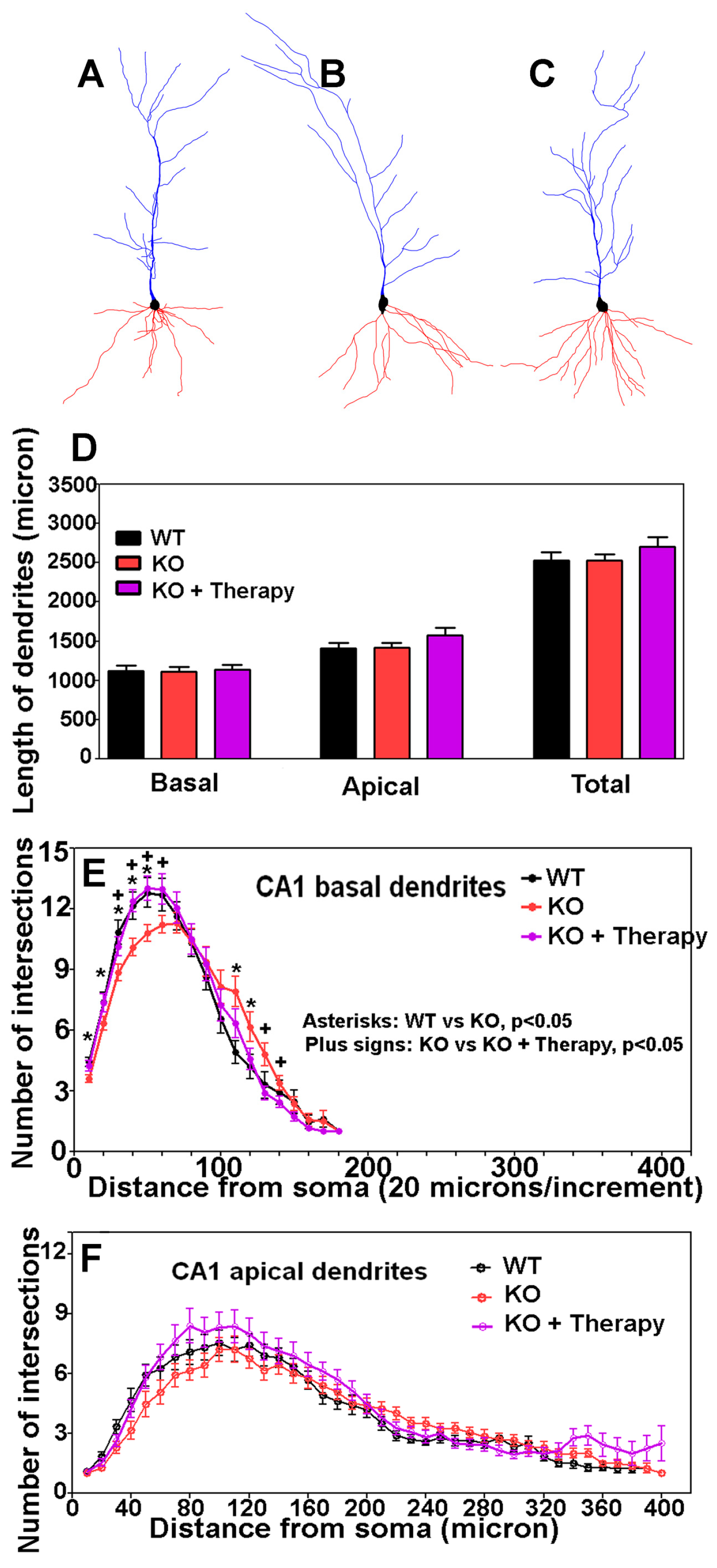
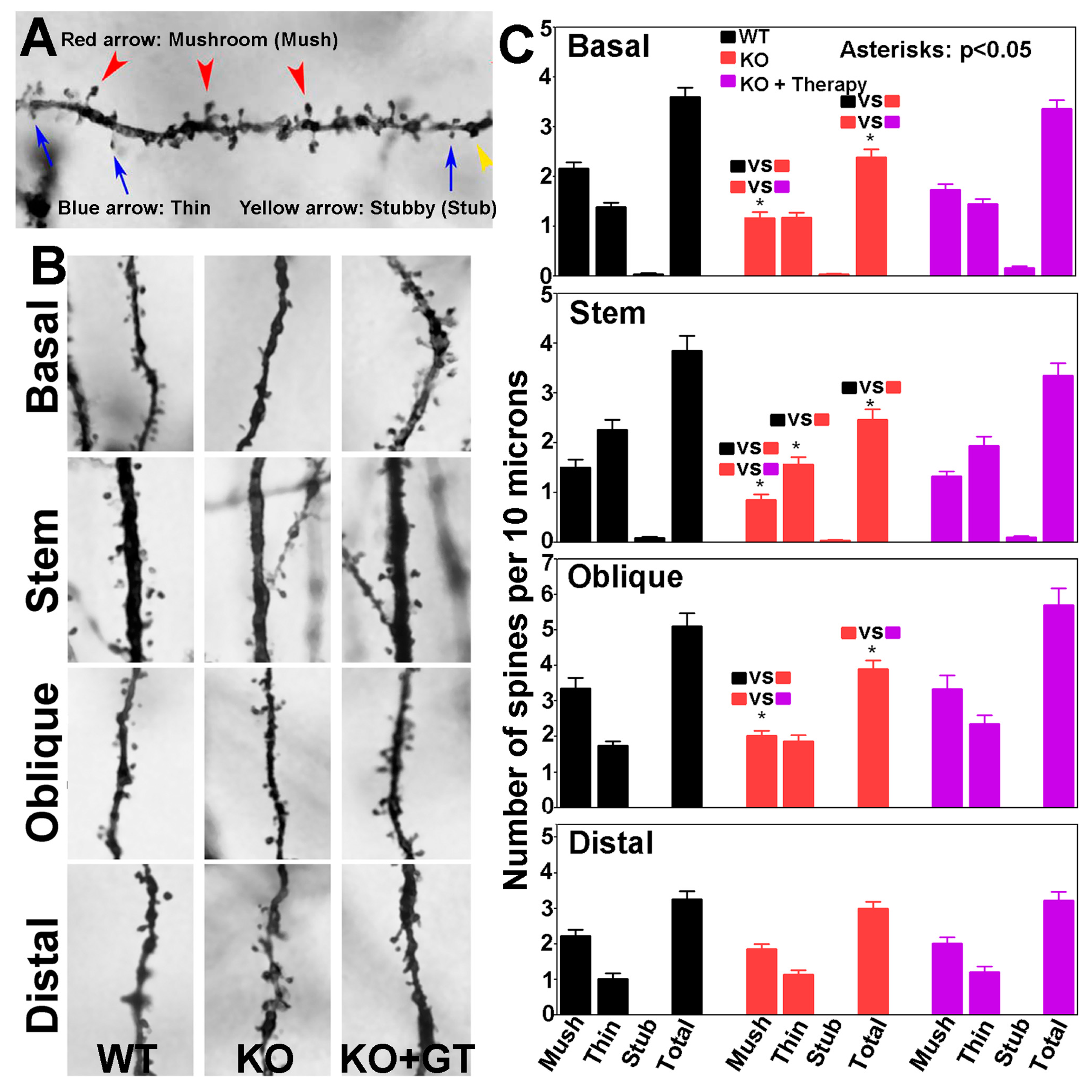
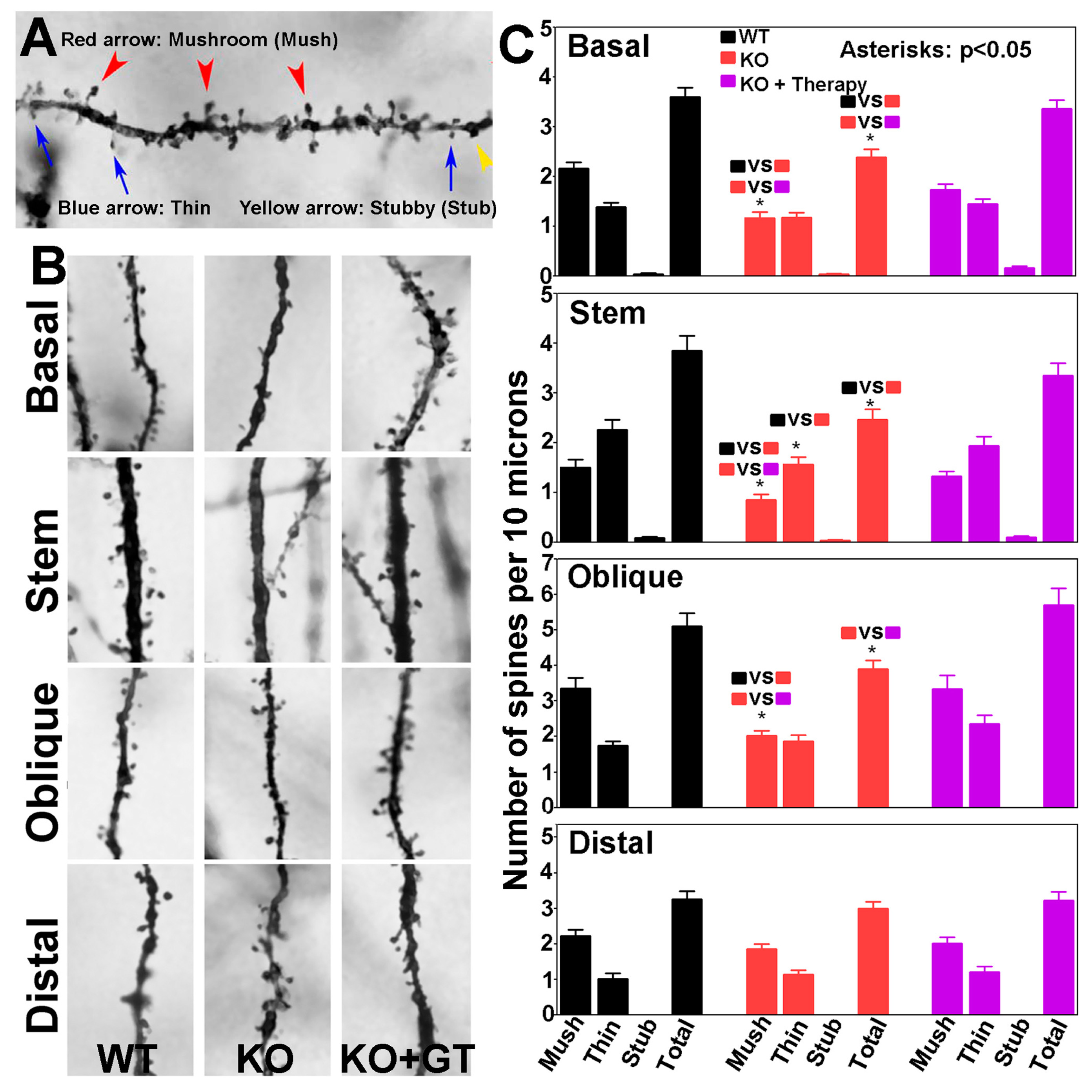
2.7. Analysis of Dendrites and Spines
2.8. Statistical Analysis
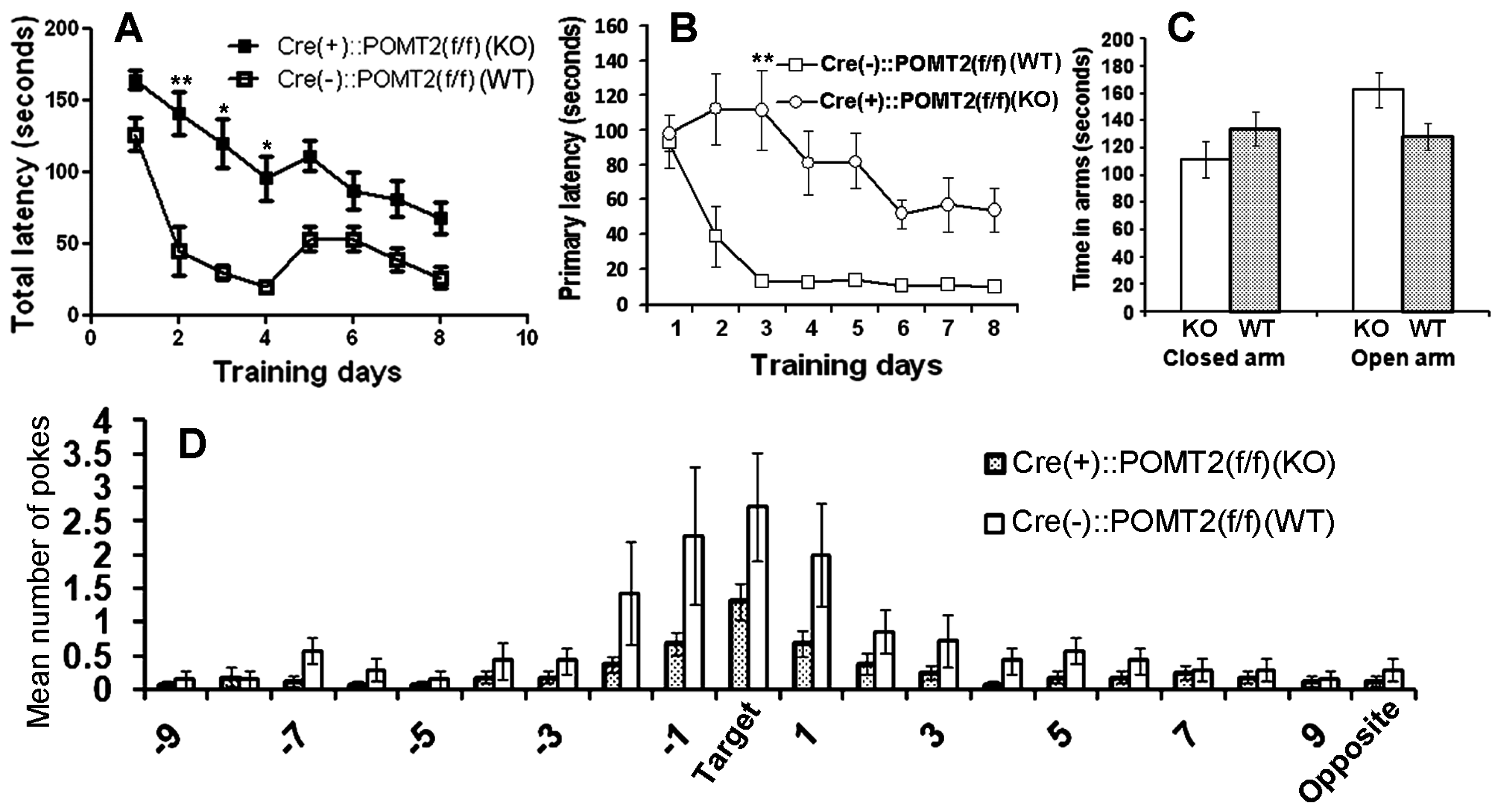
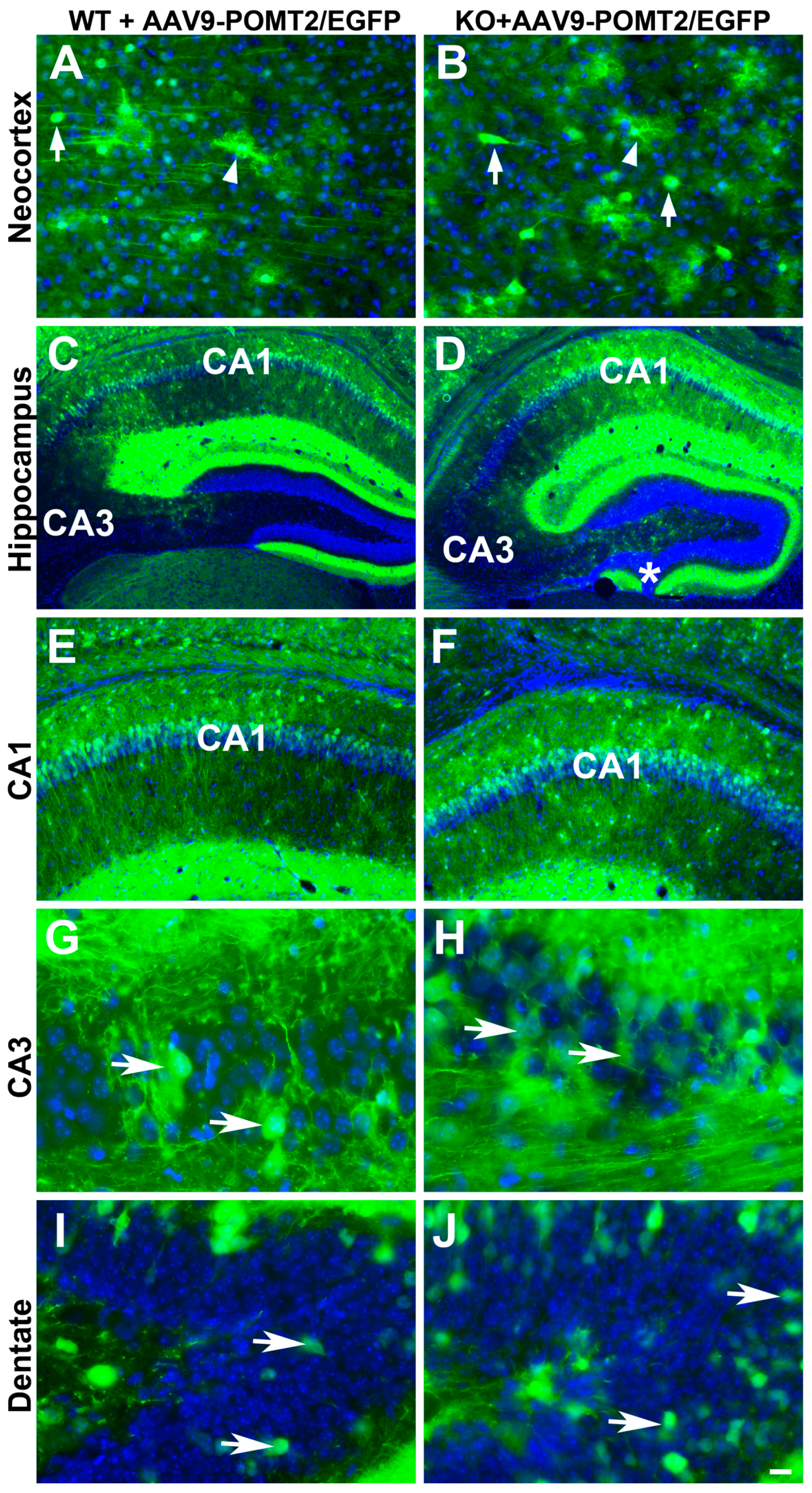
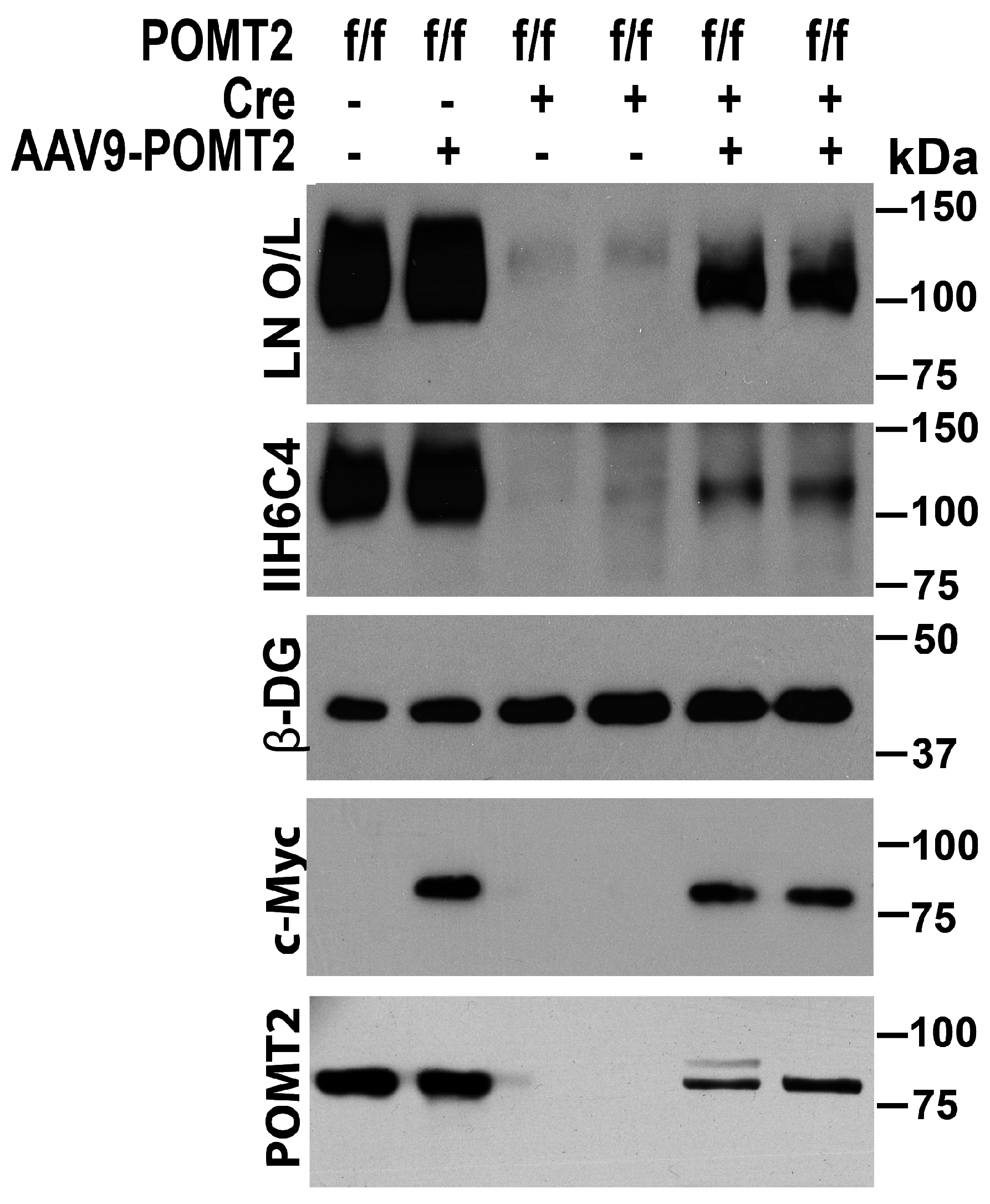
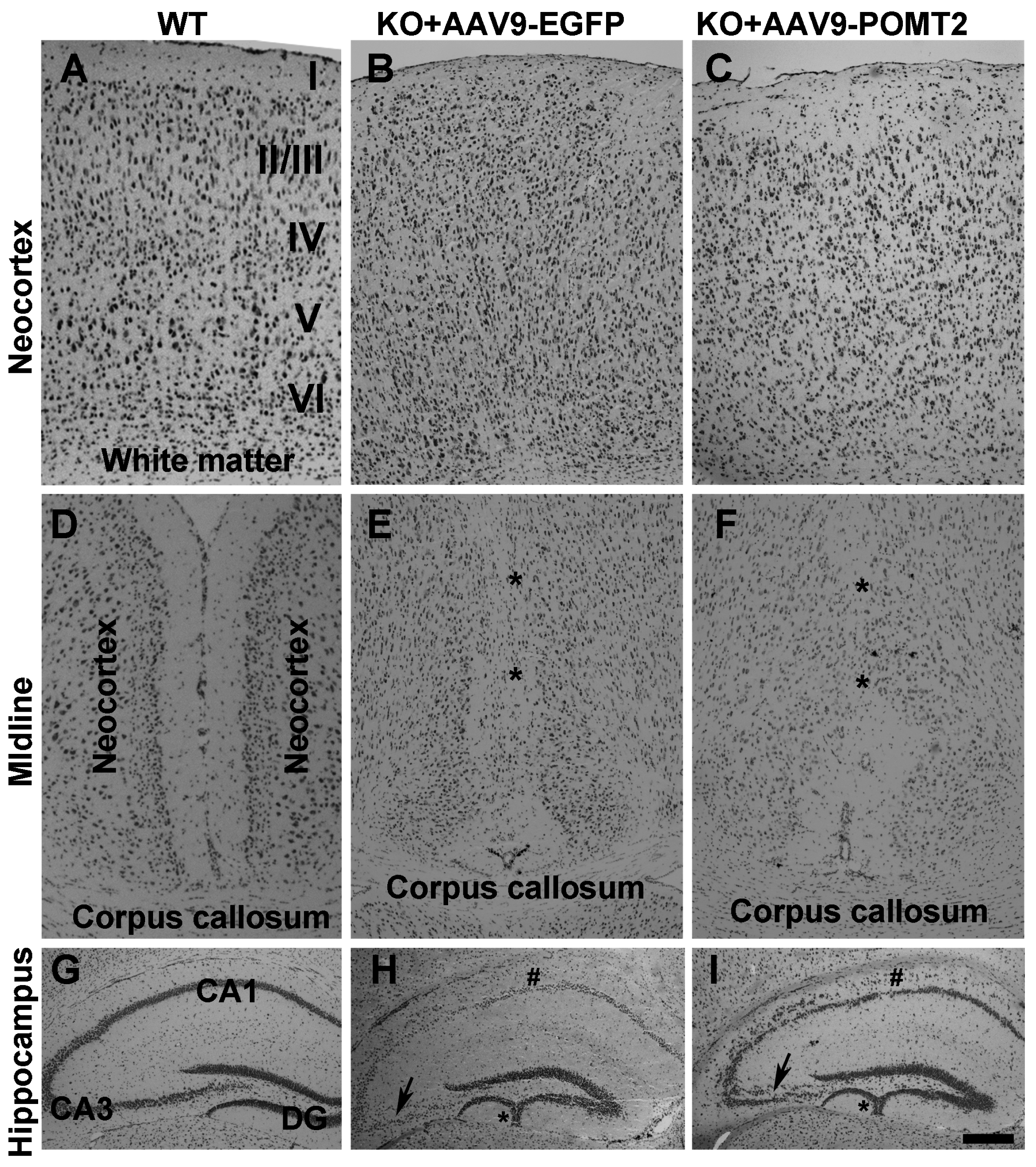
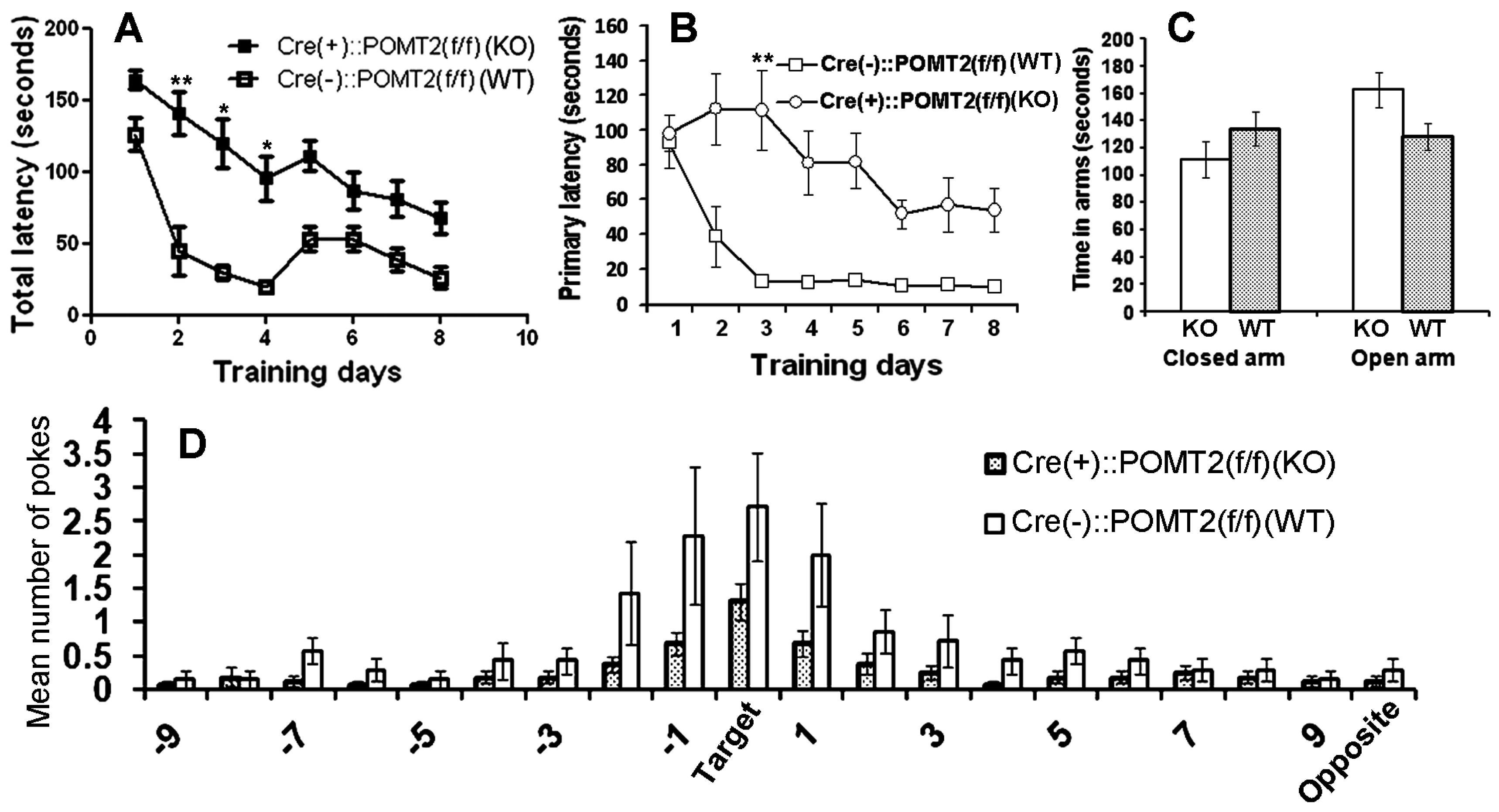
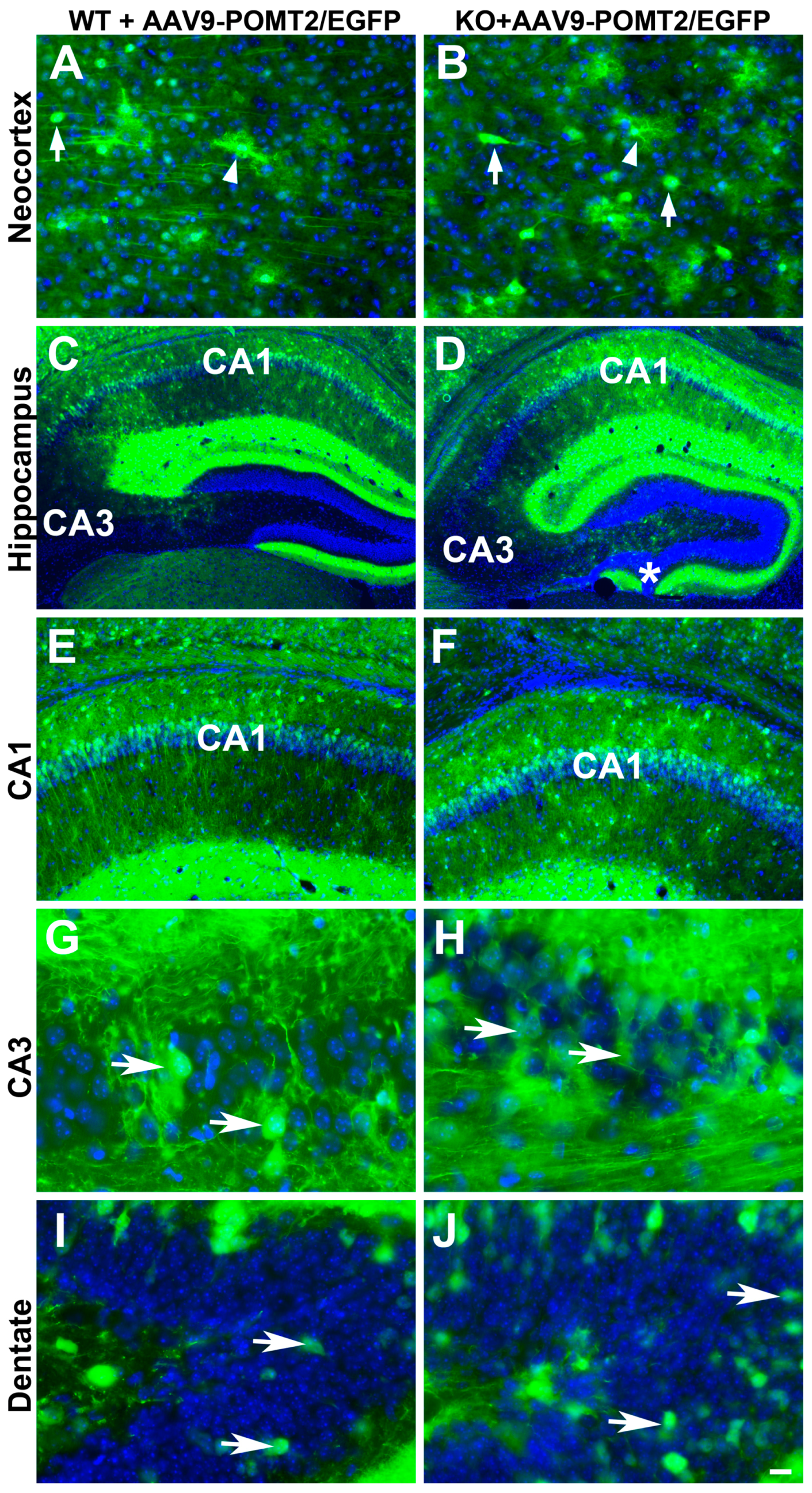
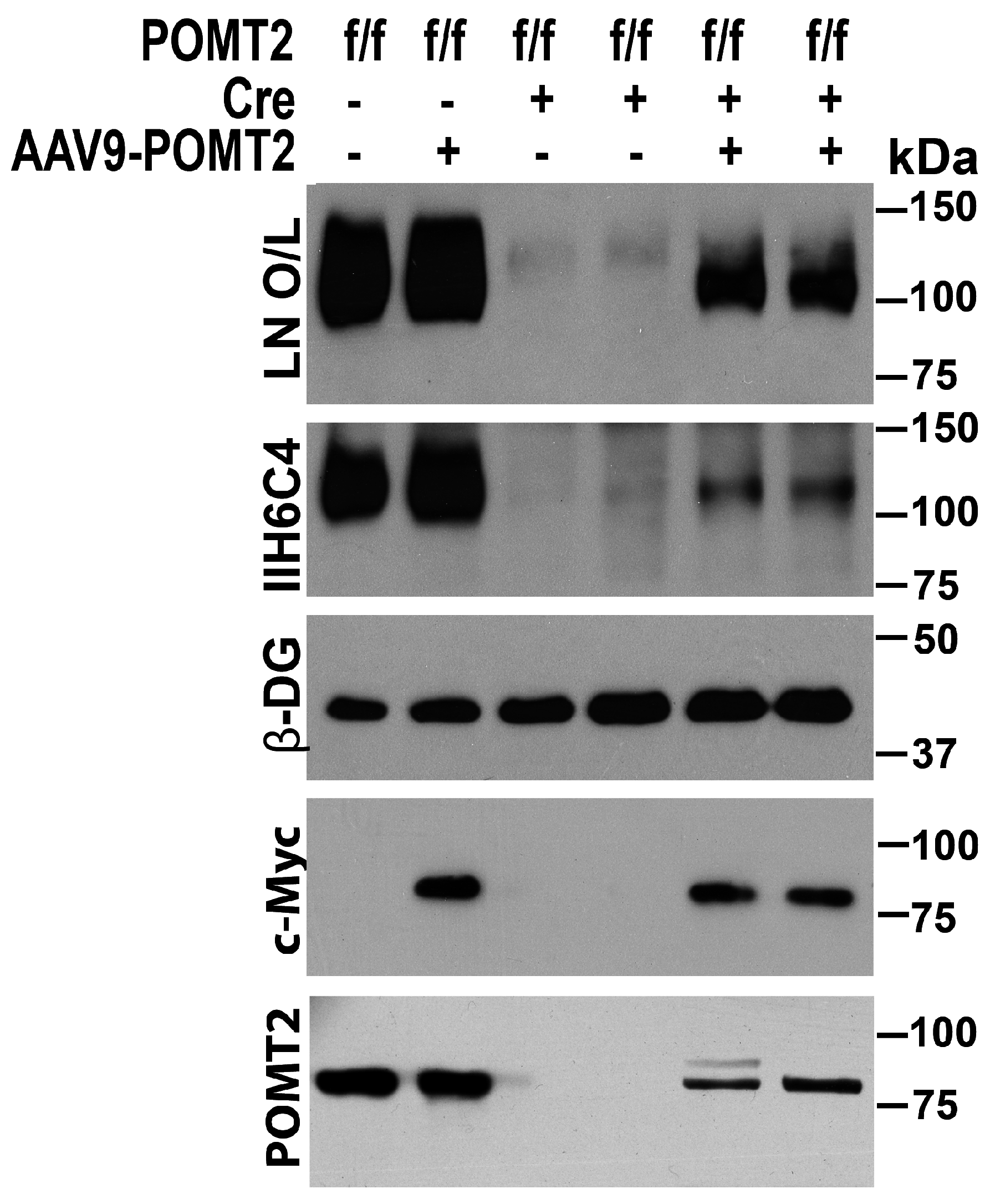
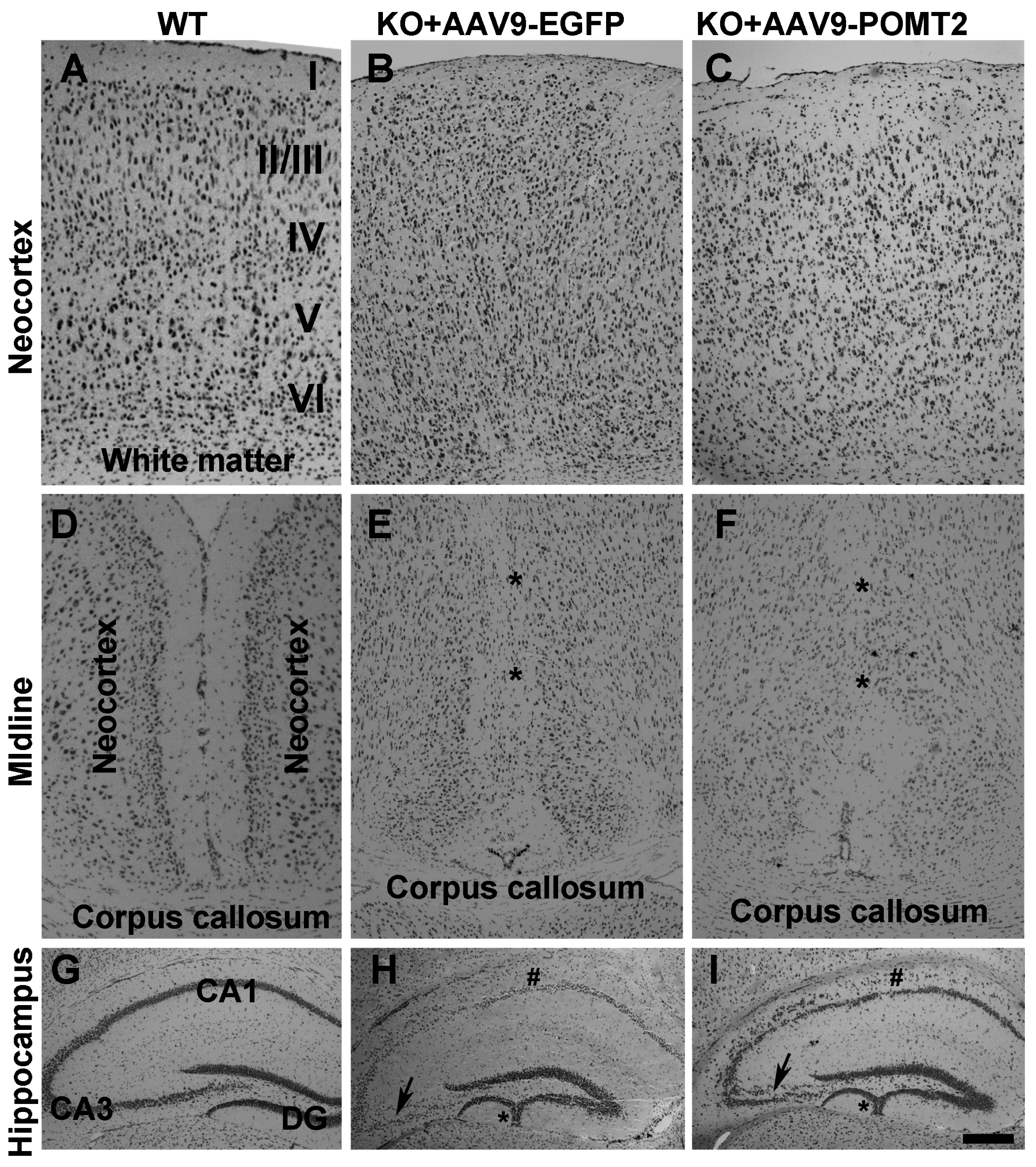
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AAV | adeno-associated viral vector |
| DG | dystroglycan |
| EGFP | enhanced green fluorescence protein |
| FKRP | fukutin-related protein |
| GT | gene therapy |
| LARGE | like-glycosyl transferase |
| PBS | phosphate-buffered saline |
| POMGnT1 | protein O-mannose N-acetylglucosaminyl transferase 1 |
| POMT2 | protein O-mannosyl transferase 2 |
| PVDF | polyvinylidene fluoride |
| TBST | Tris-buffered saline |
| WFA | wisteria floribunda lectin |
| WGA | wheat germ agglutinin |
References
- Beltrán-Valero de Bernabé, D.; Currier, S.; Steinbrecher, A.; Celli, J.; van Beusekom, E.; van der Zwaag, B.; Kayserili, H.; Merlini, L.; Chitayat, D.; Dobyns, W.B.; et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 2002, 71, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Currier, S.C.; Lee, C.K.; Chang, B.S.; Bodell, A.L.; Pai, G.S.; Job, L.; Lagae, L.G.; Al-Gazali, L.I.; Eyaid, W.M.; Enns, G.; et al. Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am. J. Med. Genet. A 2005, 133, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Van Reeuwijk, J.; Janssen, M.; van den Elzen, C.; Beltran-Valero de Bernabé, D.; Sabatelli, P.; Merlini, L.; Boon, M.; Scheffer, H.; Brockington, M.; Muntoni, F.; et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005, 42, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kobayashi, K.; Manya, H.; Taniguchi, K.; Kano, H.; Mizuno, M.; Inazu, T.; Mitsuhashi, H.; Takahashi, S.; Takeuchi, M.; et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 2001, 1, 717–724. [Google Scholar] [CrossRef]
- Longman, C.; Brockington, M.; Torelli, S.; Jimenez-Mallebrera, C.; Kennedy, C.; Khalil, N.; Feng, L.; Saran, R.K.; Voit, T.; Merlini, L.; et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum. Mol. Genet. 2003, 12, 2853–2861. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef] [PubMed]
- De Bernabé, D.B.; van Bokhoven, H.; van Beusekom, E.; Van den Akker, W.; Kant, S.; Dobyns, W.B.; Cormand, B.; Currier, S.; Hamel, B.; Talim, B.; et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J. Med. Genet. 2003, 40, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.; Bashir, R.; Burgunder, J.M.; et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero de Bernabé, D.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004, 41, e61. [Google Scholar] [CrossRef] [PubMed]
- Manzini, M.C.; Tambunan, D.E.; Hill, R.S.; Yu, T.W.; Maynard, T.M.; Heinzen, E.L.; Shianna, K.V.; Stevens, C.R.; Partlow, J.N.; Barry, B.J.; et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 2012, 91, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Buysse, K.; Riemersma, M.; Powell, G.; van Reeuwijk, J.; Chitayat, D.; Roscioli, T.; Kamsteeg, E.J.; van den Elzen, C.; van Beusekom, E.; Blaser, S.; et al. Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet. 2013, 22, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Stevens, E.; Carss, K.J.; Cirak, S.; Foley, A.R.; Torelli, S.; Willer, T.; Tambunan, D.E.; Yau, S.; Brodd, L.; Sewry, C.A.; et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2013, 92, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Roscioli, T.; Kamsteeg, E.J.; Buysse, K.; Maystadt, I.; van Reeuwijk, J.; van den Elzen, C.; van Beusekom, E.; Riemersma, M.; Pfundt, R.; Vissers, L.E.; et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of alpha-dystroglycan. Nat. Genet. 2012, 44, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Willer, T.; Lee, H.; Lommel, M.; Yoshida-Moriguchi, T.; de Bernabe, D.B.; Venzke, D.; Cirak, S.; Schachter, H.; Vajsar, J.; Voit, T.; et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012, 44, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, H. Differential glycosylation of α-dystroglycan and proteins other than α-dystroglycan by LARGE. Glycobiology 2012, 22, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Manya, H.; Chiba, A.; Yoshida, A.; Wang, X.; Chiba, Y.; Jigami, Y.; Margolis, R.U.; Endo, T. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. USA 2004, 101, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Inamori, K.; Yoshida-Moriguchi, T.; Hara, Y.; Anderson, M.E.; Yu, L.; Campbell, K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 2012, 335, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Wells, D.J.; Brown, S.C. Muscular dystrophies due to glycosylation defects: Diagnosis and therapeutic strategies. Curr. Opin. Neurol. 2011, 24, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Nishimoto, A.; Chiyonobu, T.; Takeda, S.; Miyagoe-Suzuki, Y.; Wang, F.; Fujikake, N.; Taniguchi, M.; Lu, Z.; Tachikawa, M.; et al. Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy. Hum. Mol. Genet. 2009, 18, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, C.; Fernandez-Fuente, M.; Booler, H.; Parr, C.; Kavishwar, M.; Ashraf, A.; Lacey, E.; Kim, J.; Terry, R.; Ackroyd, M.R.; et al. The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Hum. Mol. Genet. 2014, 23, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Saito, F.; Kanagawa, M.; Ikeda, M.; Hagiwara, H.; Masaki, T.; Ohkuma, H.; Katanosaka, Y.; Shimizu, T.; Sonoo, M.; Toda, T.; et al. Overexpression of LARGE suppresses muscle regeneration via down-regulation of insulin-like growth factor 1 and aggravates muscular dystrophy in mice. Hum. Mol. Genet. 2014, 23, 4543–4558. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; He, Y.; Wang, K.; Zhang, P.; Zhang, S.; Hu, H. Adeno-associated viral-mediated LARGE gene therapy rescues the muscular dystrophic phenotype in mouse models of dystroglycanopathy. Hum. Gene Ther. 2013, 24, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lu, P.J.; Wang, C.H.; Keramaris, E.; Qiao, C.; Xiao, B.; Blake, D.J.; Xiao, X.; Lu, Q.L. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha-dystroglycan and improves muscle functions. Mol. Ther. 2013, 21, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Vannoy, C.; Xu, L.; Keramaris, E.; Lu, P.; Xiao, X.; Lu, Q. AAV-mediated overexpression of LARGE rescues alpha-dystroglycan function in dystrophic mice with mutations in the fukutin-related protein. Hum. Gene Ther. Methods 2014, 25, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.J.; Xu, R.; Martin, P.T. B4GALNT2 (GALGT2) gene therapy reduces skeletal muscle pathology in the FKRP P448L mouse model of limb girdle muscular dystrophy 2I. Am. J. Pathol. 2016, 186, 2429–2448. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Wang, C.H.; Zhao, C.; Lu, P.; Awano, H.; Xiao, B.; Li, J.; Yuan, Z.; Dai, Y.; Martin, C.B.; et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol. Ther. 2014, 22, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Kanagawa, M.; Yu, C.C.; Ito, C.; Chiyo, T.; Kobayashi, K.; Okada, T.; Takeda, S.; Toda, T. Fukutin is prerequisite to ameliorate muscular dystrophic phenotype by myofiber-selective LARGE expression. Sci. Rep. 2015, 5, 8316. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, J.; Gagen, C.S.; Gray, N.W.; Zhang, Z.; Qi, Y.; Zhang, P. Conditional knockout of protein O-mannosyltransferase 2 reveals tissue specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 2011, 519, 1320–1337. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yang, Y.; Eade, A.; Xiong, Y.; Qi, Y. Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J. Comp. Neurol. 2007, 501, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, M.; Feng, G.; Hu, H.; Li, X. Breaches of the pial basement membrane are associated with defective dentate gyrus development in mouse models of congenital muscular dystrophies. Neurosci. Lett. 2011, 505, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Chiyonobu, T.; Sasaki, J.; Nagai, Y.; Takeda, S.; Funakoshi, H.; Nakamura, T.; Sugimoto, T.; Toda, T. Effects of fukutin deficiency in the developing mouse brain. Neuromuscul. Disord. 2005, 15, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.A.; Talley, T.; Qiu, M.; Puelles, L.; Rubenstein, J.L.; Jones, K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002, 22, 6309–6314. [Google Scholar] [PubMed]
- Sterio, D.C. The unbiased estimation of number and sizes of arbitrary particles using the disector. J. Microsc. 1984, 134, 127–136. [Google Scholar] [CrossRef] [PubMed]
- West, M.J. Stereological methods for estimating the total number of neurons and synapses: Issues of precision and bias. Trends Neurosci. 1999, 22, 51–61. [Google Scholar] [CrossRef]
- Sholl, D.A. The organization of the visual cortex in the cat. J. Anat. 1955, 89, 33–46. [Google Scholar] [PubMed]
- Harris, K.M.; Kater, S.B. Dendritic spines: Cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 1994, 17, 341–371. [Google Scholar] [CrossRef] [PubMed]
- Hering, H.; Sheng, M. Dendritic spines: Structure, dynamics and regulation. Nat. Rev. Neurosci. 2001, 2, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Sunyer, B.; Patil, S.; Hoger, H.; Lubec, G. Barnes maze, a useful task to assess spatial reference memory in the mice. Protoc. Exch. 2007. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Shimizu, T.; Tanaka, T.; Campbell, K.P.; Matsumura, K. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 1994, 352, 49–53. [Google Scholar] [CrossRef]
- Gee, S.H.; Blacher, R.W.; Douville, P.J.; Provost, P.R.; Yurchenco, P.D.; Carbonetto, S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J. Biol. Chem. 1993, 268, 14972–14980. [Google Scholar] [PubMed]
- Gee, S.H.; Montanaro, F.; Lindenbaum, M.H.; Carbonetto, S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell 1994, 77, 675–686. [Google Scholar] [CrossRef]
- Peng, H.B.; Ali, A.A.; Daggett, D.F.; Rauvala, H.; Hassell, J.R.; Smalheiser, N.R. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes. Commun. 1998, 5, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Sudhof, T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001, 154, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Lyon, K.A.; Leung, H.; Leahy, D.J.; Ma, L.; Ginty, D.D. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron 2012, 76, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Omori, Y.; Katoh, K.; Kondo, M.; Kanagawa, M.; Miyata, K.; Funabiki, K.; Koyasu, T.; Kajimura, N.; Miyoshi, T.; et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat. Neurosci. 2008, 11, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Omori, Y.; Sato, S.; Kobayashi, K.; Miyagoe-Suzuki, Y.; Takeda, S.; Endo, T.; Furukawa, T.; Toda, T. Post-translational maturation of dystroglycan is necessary for pikachurin binding and ribbon synaptic localization. J. Biol. Chem. 2010, 285, 31208–31216. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, J.; Zhang, Z.; Yu, M. Pikachurin interaction with dystroglycan is diminished by defective O-mannosyl glycosylation in congenital muscular dystrophy models and rescued by LARGE overexpression. Neurosci. Lett. 2011, 489, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Dino, M.R.; Harroch, S.; Hockfield, S.; Matthews, R.T. Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience 2006, 142, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.A.; Baker, E.; Hu, H.; Matthews, R.T. RPTPzeta/phosphacan is abnormally glycosylated in a model of muscle-eye-brain disease lacking functional POMGnT1. Neuroscience 2012, 220, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.; Pace, D.; Brady, L.; Gerhart, M.; Balland, A. Characterization of a novel modification on IgG2 light chain. Evidence for the presence of O-linked mannosylation. J. Chromatogr. A 2007, 1156, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Bleckmann, C.; Geyer, H.; Lieberoth, A.; Splittstoesser, F.; Liu, Y.; Feizi, T.; Schachner, M.; Kleene, R.; Reinhold, V.; Geyer, R. O-glycosylation pattern of CD24 from mouse brain. Biol. Chem. 2009, 390, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Pacharra, S.; Hanisch, F.G.; Breloy, I. Neurofascin 186 is O-mannosylated within and outside of the mucin domain. J. Proteome Res. 2012, 11, 3955–3964. [Google Scholar] [CrossRef] [PubMed]
- Vester-Christensen, M.B.; Halim, A.; Joshi, H.J.; Steentoft, C.; Bennett, E.P.; Levery, S.B.; Vakhrushev, S.Y.; Clausen, H. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc. Natl. Acad. Sci. USA 2013, 110, 21018–21023. [Google Scholar] [CrossRef] [PubMed]
- Winterhalter, P.R.; Lommel, M.; Ruppert, T.; Strahl, S. O-glycosylation of the non-canonical T-cadherin from rabbit skeletal muscle by single mannose residues. FEBS Lett. 2013, 587, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.A.; Saito, F.; Chen, J.; Michele, D.E.; Henry, M.D.; Messing, A.; Cohn, R.D.; Ross-Barta, S.E.; Westra, S.; Williamson, R.A.; et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002, 418, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Sun, M.; Liu, L.; Chen, F.; Wei, L.; Xie, W. Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 2007, 581, 2509–2516. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.B.; Li, H.L.; Kassabov, S.R.; Jin, I.; Puthanveettil, S.V.; Karl, K.A.; Lu, Y.; Kim, J.H.; Bailey, C.H.; Kandel, E.R. Neurexin-neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in aplysia. Neuron 2011, 70, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Fruh, S.; Romanos, J.; Panzanelli, P.; Burgisser, D.; Tyagarajan, S.K.; Campbell, K.P.; Santello, M.; Fritschy, J.M. Neuronal dystroglycan is necessary for formation and maintenance of functional CCK-positive basket cell terminals on pyramidal cells. J. Neurosci. 2016, 36, 10296–10313. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
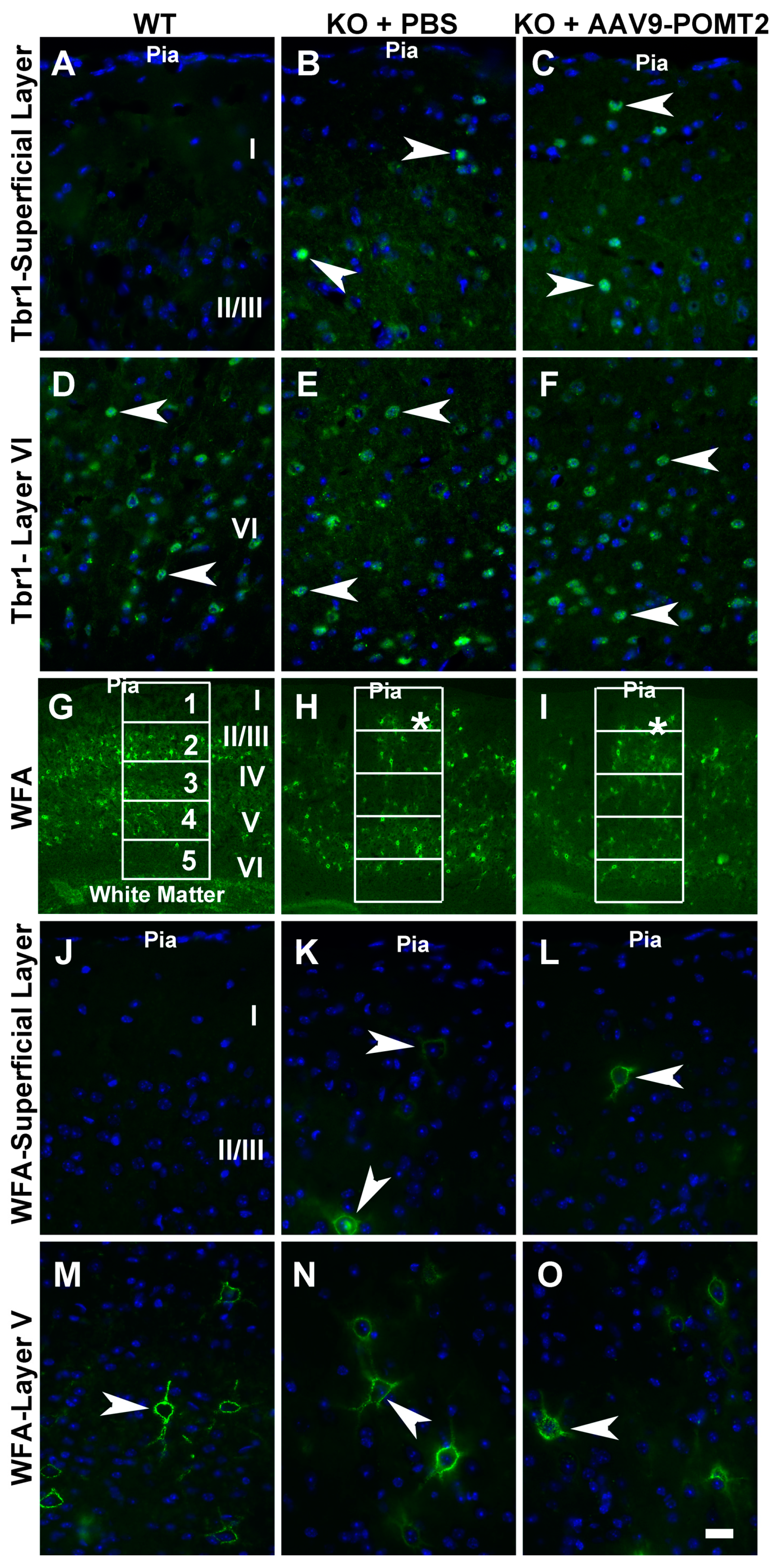
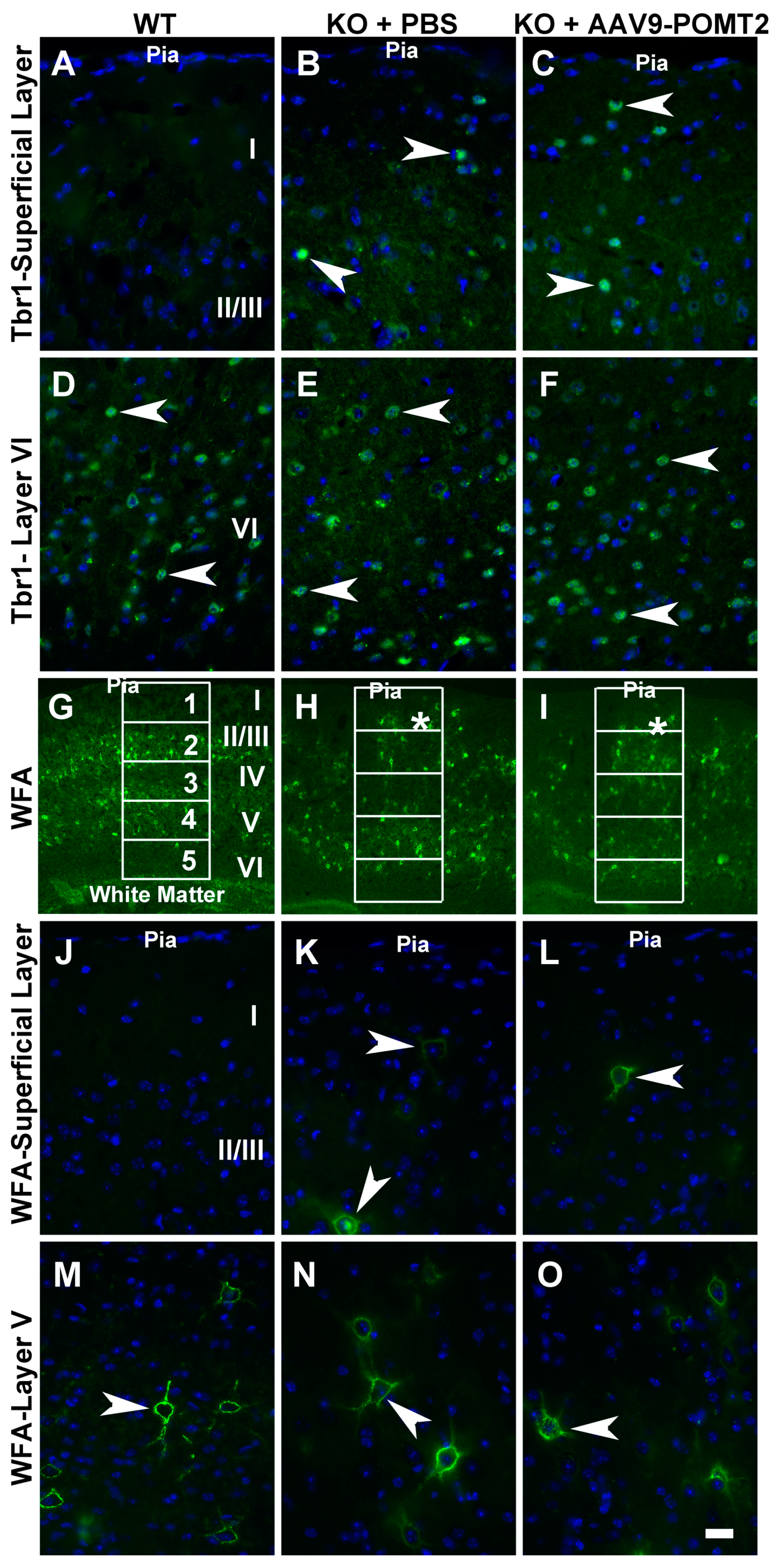
| Genotype | WFA-Labeled Neurons in Compartments 1–5 (Mean ± SEM) (see Figure 5G–I) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Wildtype | 0.8 ± 0.23 | 19 ± 3.57 | 13.2 ± 2.59 | 12.8 ± 2.59 | 3.2 ± 0.78 |
| KO + PBS | 4.2 ± 1.09 * | 17.4 ± 3.31 | 11.4 ± 2.15 | 12.4 ± 2.39 | 4.6 ± 1.18 |
| KO + AAV9-POMT2 | 4.4 ± 1.35 ** # | 16.2 ± 3.11 | 11.8 ± 2.28 | 12.0 ± 2.42 | 4.8 ± 1.12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Liu, Y.; Bampoe, K.; He, Y.; Yu, M. Postnatal Gene Therapy Improves Spatial Learning Despite the Presence of Neuronal Ectopia in a Model of Neuronal Migration Disorder. Genes 2016, 7, 105. https://doi.org/10.3390/genes7120105
Hu H, Liu Y, Bampoe K, He Y, Yu M. Postnatal Gene Therapy Improves Spatial Learning Despite the Presence of Neuronal Ectopia in a Model of Neuronal Migration Disorder. Genes. 2016; 7(12):105. https://doi.org/10.3390/genes7120105
Chicago/Turabian StyleHu, Huaiyu, Yu Liu, Kevin Bampoe, Yonglin He, and Miao Yu. 2016. "Postnatal Gene Therapy Improves Spatial Learning Despite the Presence of Neuronal Ectopia in a Model of Neuronal Migration Disorder" Genes 7, no. 12: 105. https://doi.org/10.3390/genes7120105
APA StyleHu, H., Liu, Y., Bampoe, K., He, Y., & Yu, M. (2016). Postnatal Gene Therapy Improves Spatial Learning Despite the Presence of Neuronal Ectopia in a Model of Neuronal Migration Disorder. Genes, 7(12), 105. https://doi.org/10.3390/genes7120105