
Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient


Abstract
:1. Introduction
2. Materials and Methods
3. Results
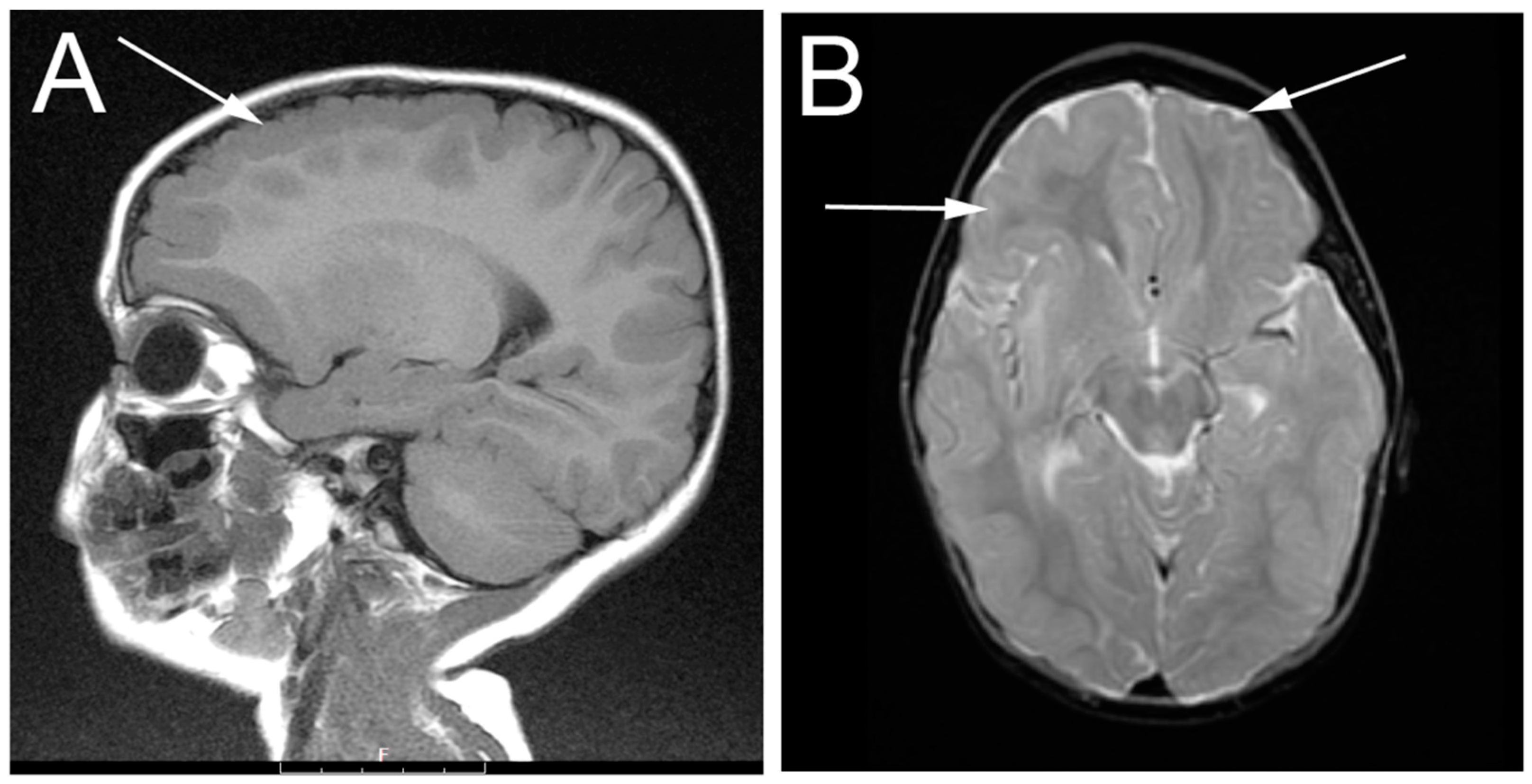
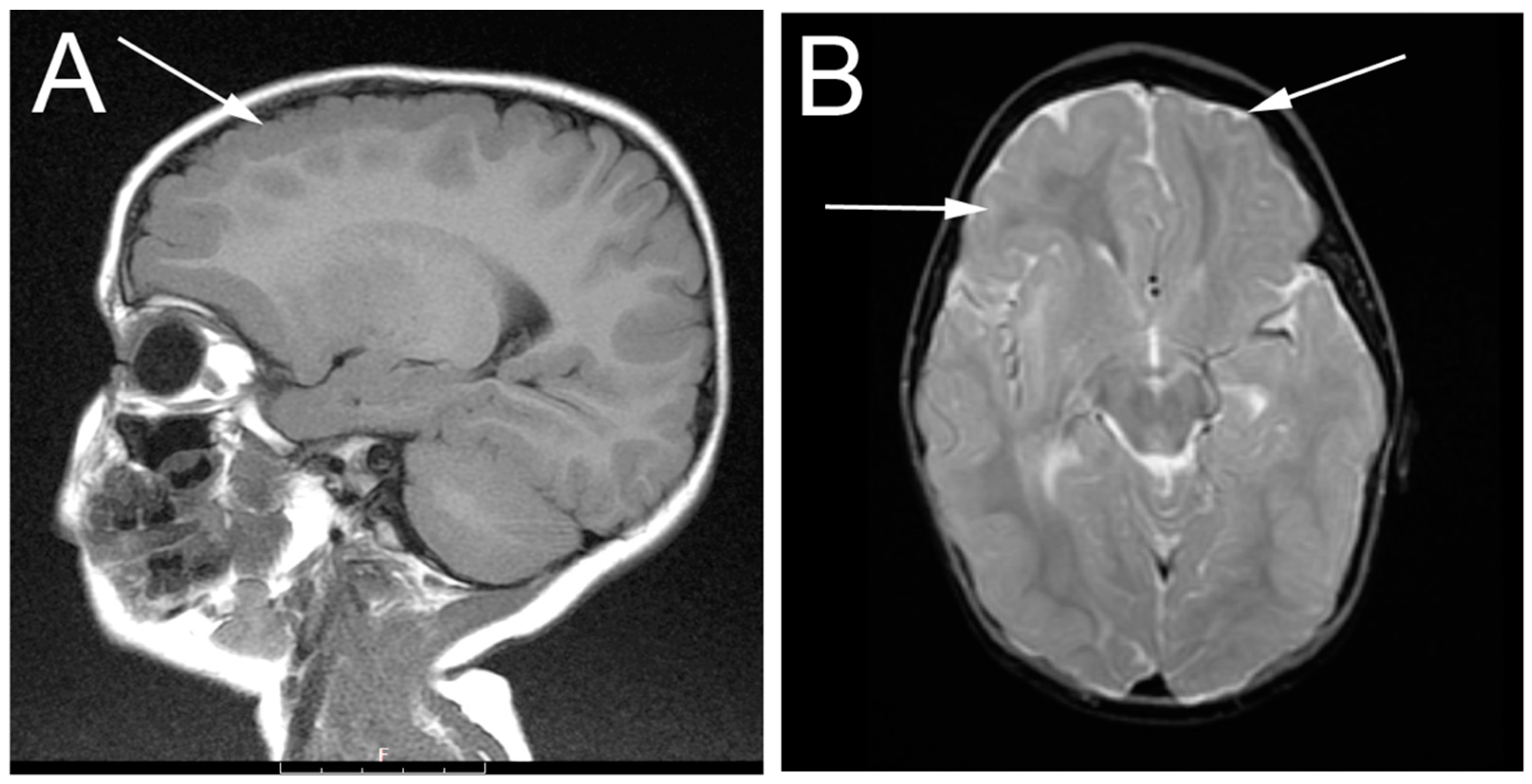
3.1. Patient Description
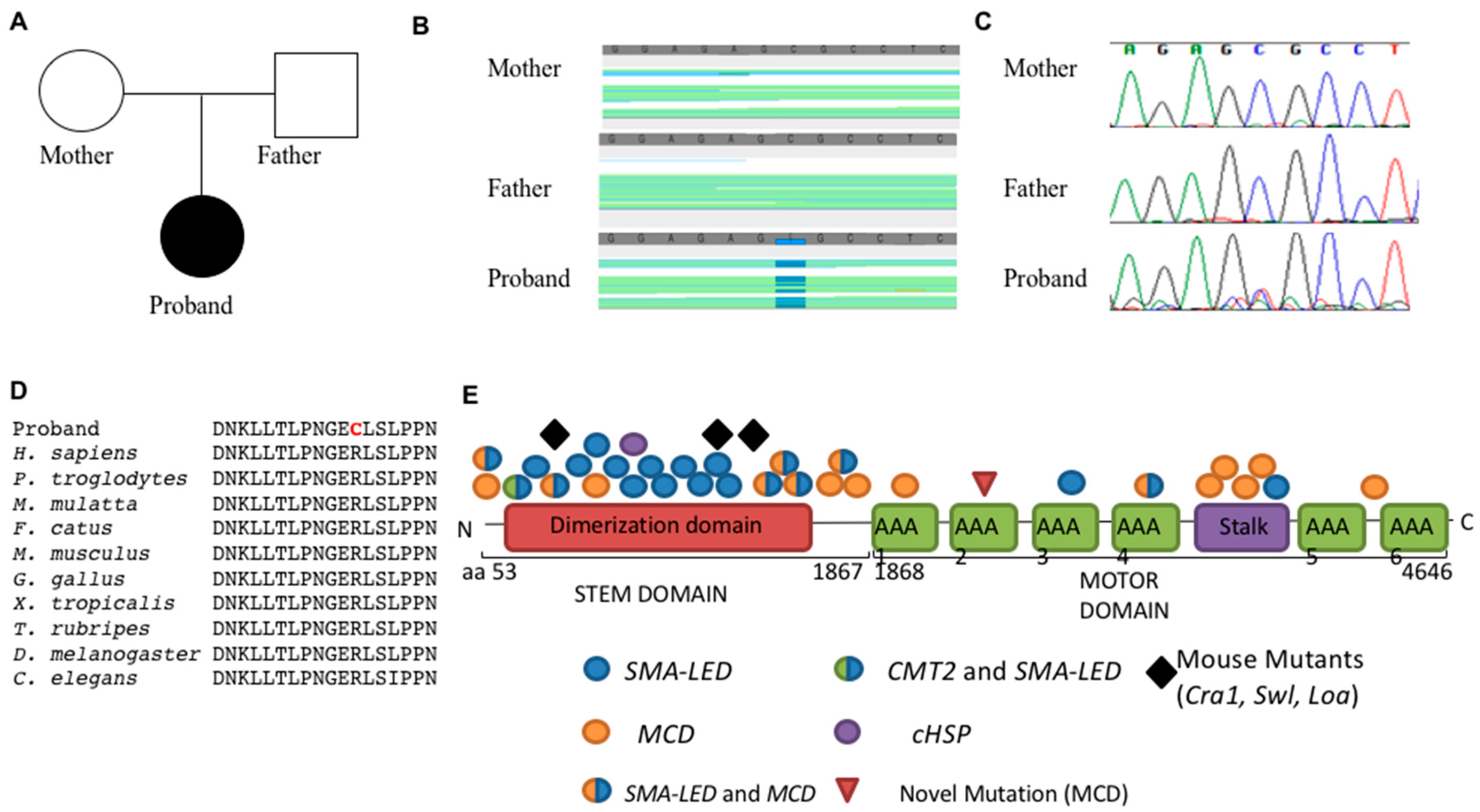
3.2. Genetics
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CCHMC | Cincinnati Children’s Hospital Medical Center |
| cHSP | Hereditary Spastic Paraplegia |
| CMT2 | Charcot-Marie-Tooth Type 2 |
| DYNC1H1 | cytoplasmic dynein heavy chain 1 |
| EEG | Electroencephalogram |
| EXAC | Exome aggregation consortium |
| GATK | Genome Analysis Toolkit |
| MCD | malformations of cortical development |
| MRI | Magnetic Resonance Imaging |
| PAFAH1B1 | platelet-activating factor acetylhydrolase isoform 1B, alpha subunit |
| SMA | spinal muscular atrophy |
| SMA-LED | Spinal Muscular Atrophy-Lower Extremity Dominant |
| TORCH | Toxoplasma gondii, Other viruses, Rubella, Cytomegalovirus, and Herpes simplex |
References
- Peeters, K.; Bervoets, S.; Chamova, T.; Litvinenko, I.; de Vriendt, E.; Bichev, S.; Kancheva, D.; Mitev, V.; Kennerson, M.; Timmerman, V.; et al. Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies. Hum. Mutat. 2015, 36, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.B.; Ori-McKenney, K.M.; Scoto, M.; Tuck, E.P.; Bell, S.; Ma, D.; Masi, S.; Allred, P.; Al-Lozi, M.; Reilly, M.M.; et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012, 78, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, C.; Moro, F.; Yi, J.; Weil, S.; Brisca, G.; Astrea, G.; Severino, M.; Romano, A.; Battini, R.; Rossi, A.; et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum. Mutat. 2014, 35, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, Y.; Saitoh, S.; Tomizawa, K.; Sudo, A.; Asahina, N.; Shiraishi, H.; Ito, J.; Tanaka, H.; Doi, H.; Saitsu, H.; et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics 2012, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Strickland, A.V.; Schabhuttl, M.; Offenbacher, H.; Synofzik, M.; Hauser, N.S.; Brunner-Krainz, M.; Gruber-Sedlmayr, U.; Moore, S.A.; Windhager, R.; Bender, B.; et al. Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J. Neurol. 2015, 262, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.H.; Vissers, L.E.; Willemsen, M.A.; van Bon, B.W.; Kroes, T.; de Ligt, J.; de Vries, B.B.; Schoots, J.; Lugtenberg, D.; Hamel, B.C.; et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J. Med. Genet. 2012, 49, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Greensmith, L.; Hafezparast, M.; Fisher, E.M. Cytoplasmic dynein heavy chain: The servant of many masters. Trends Neurosci. 2013, 36, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.W.; Bremner, K.H.; Vallee, R.B. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 2007, 10, 970–979. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Petrovski, S.; Xie, P.; Ruzzo, E.K.; Lu, Y.F.; McSweeney, K.M.; Ben-Zeev, B.; Nissenkorn, A.; Anikster, Y.; Oz-Levi, D.; et al. Whole-exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Genet. Med. 2015, 17, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.K.; Wen, X.J.; Zhou, C.J. Microtubule configuration and membranous vesicle transport in elongating fiber cells of the rat lens. Exp. Eye Res. 2003, 77, 615–626. [Google Scholar] [CrossRef]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef] [PubMed]
- El-Kadi, A.M.; Bros-Facer, V.; Deng, W.; Philpott, A.; Stoddart, E.; Banks, G.; Jackson, G.S.; Fisher, E.M.; Duchen, M.R.; Greensmith, L.; et al. The legs at odd angles (Loa) mutation in cytoplasmic dynein ameliorates mitochondrial function in SOD1G93A mouse model for motor neuron disease. J. Biol. Chem. 2010, 285, 18627–18639. [Google Scholar] [PubMed]
- De Angelis, M.H.; Flaswinkel, H.; Fuchs, H.; Rathkolb, B.; Soewarto, D.; Marschall, S.; Heffner, S.; Pargent, W.; Wuensch, K.; Jung, M.; et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000, 25, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Ori-McKenney, K.M.; Vallee, R.B. Neuronal migration defects in the Loa dynein mutant mouse. Neural Dev. 2011. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Levedakou, E.N.; Millen, K.J.; Wollmann, R.L.; Soliven, B.; Popko, B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci. 2007, 27, 14515–14524. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Rossor, A.M.; Harms, M.B.; Cirak, S.; Calissano, M.; Robb, S.; Manzur, A.Y.; Martínez Arroyo, A.; Rodriguez Sanz, A.; Mansour, S.; et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology 2015, 84, 668–679. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Familial Variant Analysis | # of Variants |
|---|---|
| Total Variants | 114,284 |
| Quality Control > 20 and Read Depth > 10 | 96,646 |
| De Novo Variant Analysis | |
| Variants with MAF < 0.01 (Exac, 1000 genomes project, NHLBI ESP6500 exome data) | 13,452 |
| Coding, non-synonymous variants | 1192 |
| De novo mutations | 70 |
| Variants with alt allele > 0.3 freq. in proband | 9 |
| Variants supported by manual inspection of bam files | 1 (DYNC1H1) |
| Homozygous Recessive Analysis | |
| Variants with MAF < 0.03 (Exac, 1000 genomes project, NHLBI ESP6500 exome data) | 16,546 |
| Coding, non-synonymous variants | 1799 |
| Homozygous recessive mutations | 11 |
| Variants supported by manual inspection of bam files | 9 |
| Remove variants seen in homozygotes in Exac | 1 |
| Gene causes human disease not seen in proband | 1 |
| Compound Heterozygous Analysis | |
| Variants with MAF < 0.03 (Exac, 1000 genomes project, NHLBI ESP6500 exome data) | 16,546 |
| Coding, non-synonymous variants | 1799 |
| Genes Represented with compound heterozygous mutations | 16 |
| Remove genes for which variants are seen as homozygotes in Exac | 3 |
| Gene known to causes human disease not seen in proband | 2 |
| Known gene expression not consistent with disease in proband | 1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelineau-Morel, R.; Lukacs, M.; Weaver, K.N.; Hufnagel, R.B.; Gilbert, D.L.; Stottmann, R.W. Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient. Genes 2016, 7, 85. https://doi.org/10.3390/genes7100085
Gelineau-Morel R, Lukacs M, Weaver KN, Hufnagel RB, Gilbert DL, Stottmann RW. Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient. Genes. 2016; 7(10):85. https://doi.org/10.3390/genes7100085
Chicago/Turabian StyleGelineau-Morel, Rose, Marshall Lukacs, K. Nicole Weaver, Robert B. Hufnagel, Donald L. Gilbert, and Rolf W. Stottmann. 2016. "Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient" Genes 7, no. 10: 85. https://doi.org/10.3390/genes7100085
APA StyleGelineau-Morel, R., Lukacs, M., Weaver, K. N., Hufnagel, R. B., Gilbert, D. L., & Stottmann, R. W. (2016). Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient. Genes, 7(10), 85. https://doi.org/10.3390/genes7100085