Phenotype-Based Genetic Association Studies (PGAS)—Towards Understanding the Contribution of Common Genetic Variants to Schizophrenia Subphenotypes

Abstract
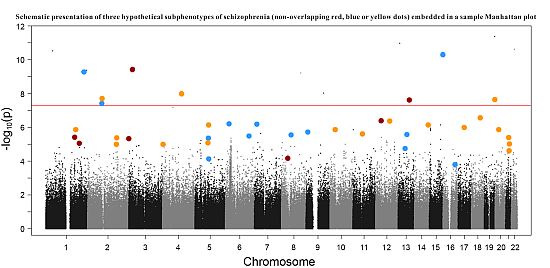
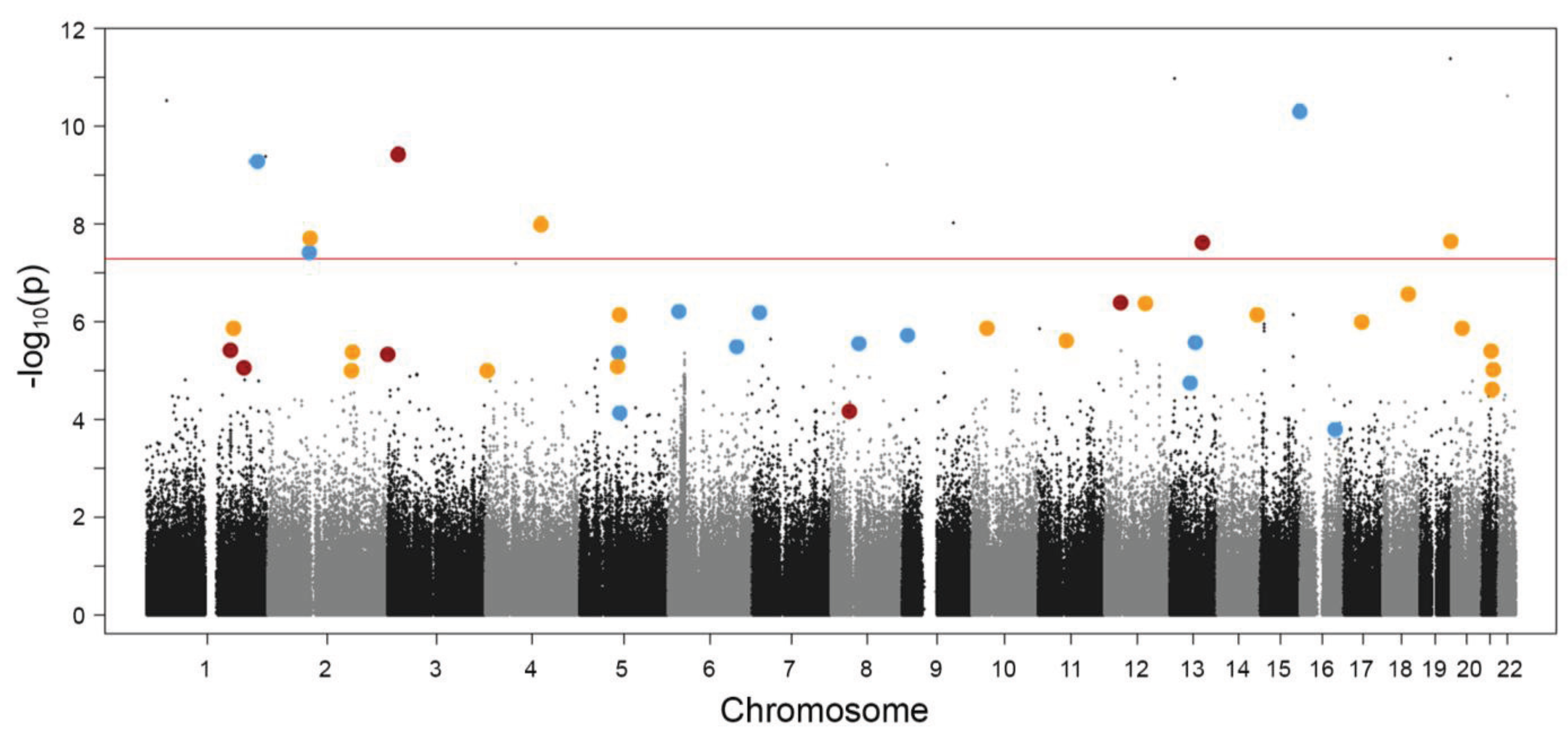
:
{kind=link}
{kind=link}
1. Schizophrenia Is a Heterogeneous Group of Diseases Diagnosed Purely Clinically
2. Despite High Heritability of Schizophrenia No “Disease Genes” Have Been Uncovered
4. Definition of Biological Subgroups of Schizophrenia: The Essential Next Step
5. Deep Phenotyping Is a Prerequisite for PGAS: The GRAS Data Collection
6. Phenotype-Based Genetic Association Studies (PGAS): Proof-of-Principle
7. PGAS Will Be Instrumental for Definition of Biological Schizophrenia Subgroups
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cardno, A.G.; Gottesman, I.I. Twin studies of schizophrenia: From bow-and-arrow concordances to star wars mx and functional genomics. Am. J. Med. Genet. 2000, 97, 12–17. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Yip, B.H.; Bjork, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic determinants of schizophrenia and bipolar disorder in swedish families: A population-based study. Lancet 2009, 373, 234–239. [Google Scholar] [CrossRef]
- Cardno, A.G.; Marshall, E.J.; Coid, B.; Macdonald, A.M.; Ribchester, T.R.; Davies, N.J.; Venturi, P.; Jones, L.A.; Lewis, S.W.; Sham, P.C.; et al. Heritability estimates for psychotic disorders: The maudsley twin psychosis series. Arch. Gen. Psychiatry 1999, 56, 162–168. [Google Scholar]
- Franzek, E.; Beckmann, H. Different genetic background of schizophrenia spectrum psychoses: A twin study. Am. J. Psychiatry 1998, 155, 76–83. [Google Scholar]
- Svrakic, D.M.; Zorumski, C.F.; Svrakic, N.M.; Zwir, I.; Cloninger, C.R. Risk architecture of schizophrenia: The role of epigenetics. Curr. Opin. Psychiatry 2013, 26, 188–195. [Google Scholar] [CrossRef]
- Van Os, J.; Kenis, G.; Rutten, B.P. The environment and schizophrenia. Nature 2010, 468, 203–212. [Google Scholar] [CrossRef]
- Giusti-Rodriguez, P.; Sullivan, P.F. The genomics of schizophrenia: Update and implications. J. Clin. Invest. 2013, 123, 4557–4563. [Google Scholar] [CrossRef]
- Sullivan, P.F. The genetics of schizophrenia. PLoS Med. 2005, 2, e212. [Google Scholar] [CrossRef]
- St. Clair, D.; Blackwood, D.; Muir, W.; Carothers, A.; Walker, M.; Spowart, G.; Gosden, C.; Evans, H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990, 336, 13–16. [Google Scholar] [CrossRef]
- Brandon, N.J.; Millar, J.K.; Korth, C.; Sive, H.; Singh, K.K.; Sawa, A. Understanding the role of disc1 in psychiatric disease and during normal development. J. Neurosci. 2009, 29, 12768–12775. [Google Scholar]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kahler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar]
- Consortium, I.S.; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar]
- Papiol, S.; Malzahn, D.; Kästner, A.; Sperling, S.; Begemann, M.; Stefansson, H.; Bickeboller, H.; Nave, K.A.; Ehrenreich, H. Dissociation of accumulated genetic risk and disease severity in patients with schizophrenia. Transl. Psychiatry 2011, 1, e45. [Google Scholar] [CrossRef]
- Papiol, S.; Mitjans, M.; Assogna, F.; Piras, F.; Hammer, C.; Caltagirone, C.; Arias, B.; Ehrenreich, H.; Spalletta, G. Polygenic determinants of white matter volume derived from gwas lack reproducibility in a replicate sample. Transl. Psychiatry 2014. [Google Scholar] [CrossRef]
- Terwisscha van Scheltinga, A.F.; Bakker, S.C.; van Haren, N.E.; Derks, E.M.; Buizer-Voskamp, J.E.; Boos, H.B.; Cahn, W.; Hulshoff Pol, H.E.; Ripke, S.; Ophoff, R.A.; et al. Genetic schizophrenia risk variants jointly modulate total brain and white matter volume. Biol. Psychiatry 2013, 73, 525–531. [Google Scholar] [CrossRef]
- Stepniak, B.; Papiol, S.; Hammer, C.; Ramin, A.; Everts, S.; Hennig, L.; Begemann, M.; Ehrenreich, H. Accumulated environmental risk determining the onset of schizophrenia. Submitted for publication. 2014. [Google Scholar]
- Begemann, M.; Grube, S.; Papiol, S.; Malzahn, D.; Krampe, H.; Ribbe, K.; Friedrichs, H.; Radyushkin, K.A.; El-Kordi, A.; Benseler, F.; et al. Modification of cognitive performance in schizophrenia by complexin 2 gene polymorphisms. Arch. Gen. Psychiatry 2010, 67, 879–888. [Google Scholar] [CrossRef]
- Ribbe, K.; Friedrichs, H.; Begemann, M.; Grube, S.; Papiol, S.; Kästner, A.; Gerchen, M.F.; Ackermann, V.; Tarami, A.; Treitz, A.; et al. The cross-sectional gras sample: A comprehensive phenotypical data collection of schizophrenic patients. BMC Psychiatry 2010, 10, 91. [Google Scholar] [CrossRef]
- Van Os, J.; Kapur, S. Schizophrenia. Lancet 2009, 374, 635–645. [Google Scholar] [CrossRef]
- Grube, S.; Gerchen, M.F.; Adamcio, B.; Pardo, L.A.; Martin, S.; Malzahn, D.; Papiol, S.; Begemann, M.; Ribbe, K.; Friedrichs, H.; et al. A cag repeat polymorphism of kcnn3 predicts sk3 channel function and cognitive performance in schizophrenia. EMBO Mol. Med. 2011, 3, 309–319. [Google Scholar] [CrossRef]
- Kästner, A.; Grube, S.; El-Kordi, A.; Stepniak, B.; Friedrichs, H.; Sargin, D.; Schwitulla, J.; Begemann, M.; Giegling, I.; Miskowiak, K.W.; et al. Common variants of the genes encoding erythropoietin and its receptor modulate cognitive performance in schizophrenia. Mol. Med. 2012, 18, 1029–1040. [Google Scholar]
- Gong, Y.G.; Wu, C.N.; Xing, Q.H.; Zhao, X.Z.; Zhu, J.; He, L. A two-method meta-analysis of neuregulin 1(nrg1) association and heterogeneity in schizophrenia. Schizophr. Res. 2009, 111, 109–114. [Google Scholar] [CrossRef]
- Li, D.; Collier, D.A.; He, L. Meta-analysis shows strong positive association of the neuregulin 1 (nrg1) gene with schizophrenia. Hum. Mol. Genet. 2006, 15, 1995–2002. [Google Scholar] [CrossRef]
- Munafo, M.R.; Thiselton, D.L.; Clark, T.G.; Flint, J. Association of the nrg1 gene and schizophrenia: A meta-analysis. Mol. Psychiatry 2006, 11, 539–546. [Google Scholar] [CrossRef]
- Papiol, S.; Begemann, M.; Rosenberger, A.; Friedrichs, H.; Ribbe, K.; Grube, S.; Schwab, M.H.; Jahn, H.; Gunkel, S.; Benseler, F.; et al. A phenotype-based genetic association study reveals the contribution of neuregulin1 gene variants to age of onset and positive symptom severity in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatry Genet. 2011, 156, 340–345. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Goebbels, S.; Papiol, S.; Kästner, A.; Hofer, S.; Begemann, M.; Gerwig, U.C.; Boretius, S.; Wieser, G.L.; Ronnenberg, A.; et al. A myelin gene causative of a catatonia-depression syndrome upon aging. EMBO Mol. Med. 2012, 4, 528–539. [Google Scholar] [CrossRef]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Siren, A.L.; Pardo, L.A.; Sperling, S.; Mohd Jofrry, S.; et al. Neuropsychiatric disease relevance of circulating anti-nmda receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 2013. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Nave, K.-A.; Max Planck Institute of Experimental Medicine, and DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), Hermann-Rein-Str.3, 37075 Göttingen, Germany. Unpublished work. 2014.
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ehrenreich, H.; Nave, K.-A. Phenotype-Based Genetic Association Studies (PGAS)—Towards Understanding the Contribution of Common Genetic Variants to Schizophrenia Subphenotypes. Genes 2014, 5, 97-105. https://doi.org/10.3390/genes5010097
Ehrenreich H, Nave K-A. Phenotype-Based Genetic Association Studies (PGAS)—Towards Understanding the Contribution of Common Genetic Variants to Schizophrenia Subphenotypes. Genes. 2014; 5(1):97-105. https://doi.org/10.3390/genes5010097
Chicago/Turabian StyleEhrenreich, Hannelore, and Klaus-Armin Nave. 2014. "Phenotype-Based Genetic Association Studies (PGAS)—Towards Understanding the Contribution of Common Genetic Variants to Schizophrenia Subphenotypes" Genes 5, no. 1: 97-105. https://doi.org/10.3390/genes5010097
APA StyleEhrenreich, H., & Nave, K.-A. (2014). Phenotype-Based Genetic Association Studies (PGAS)—Towards Understanding the Contribution of Common Genetic Variants to Schizophrenia Subphenotypes. Genes, 5(1), 97-105. https://doi.org/10.3390/genes5010097