Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
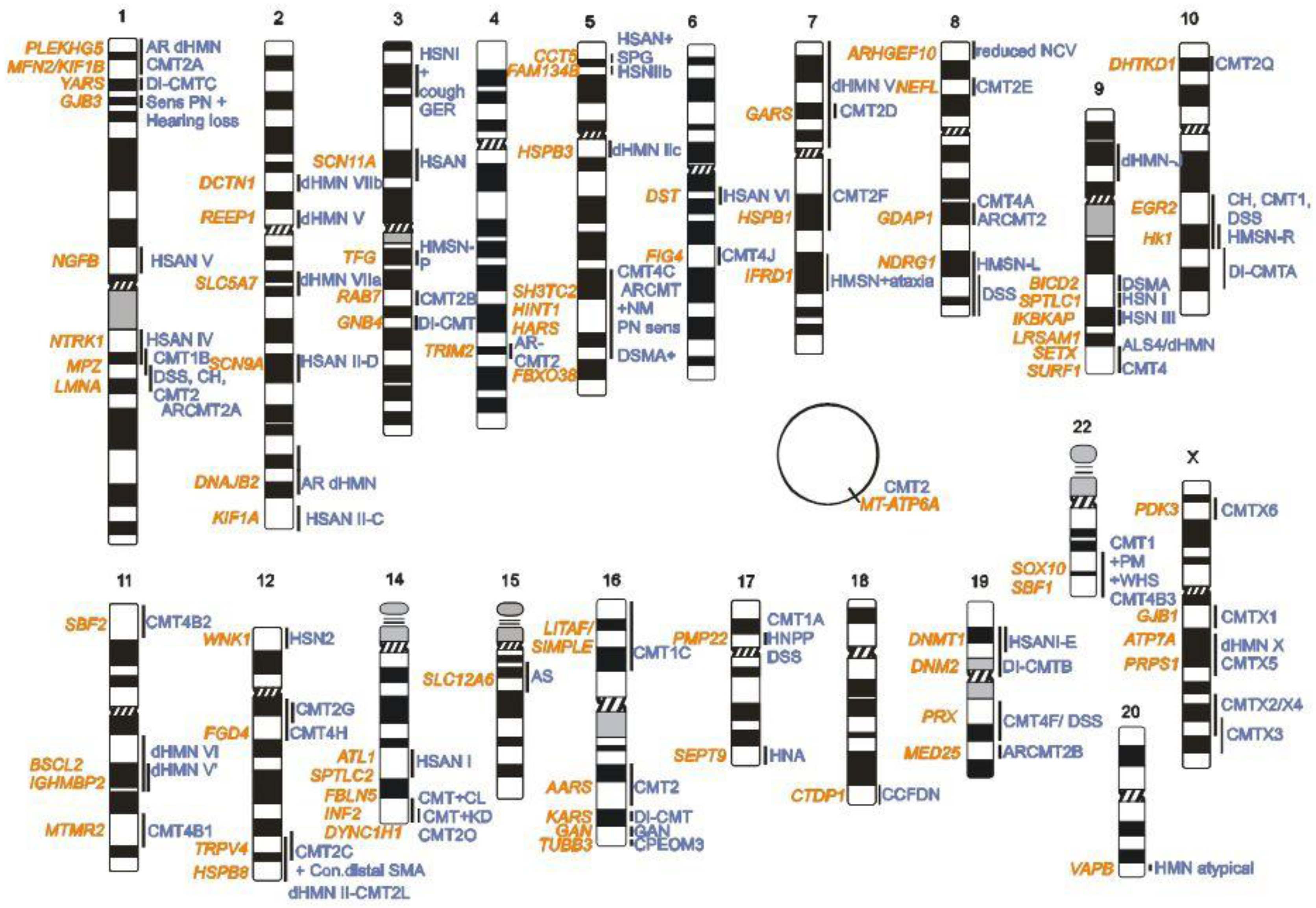
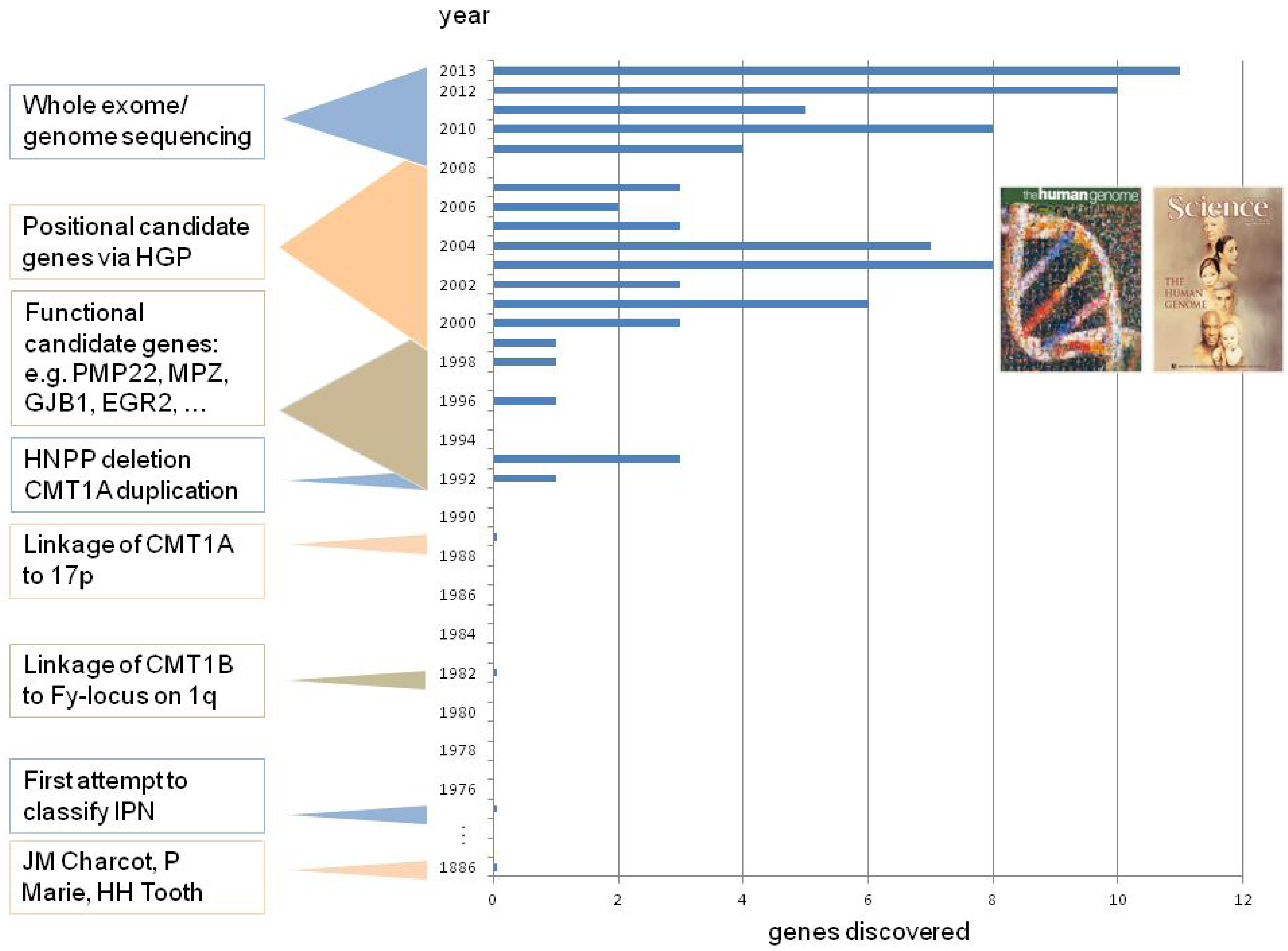
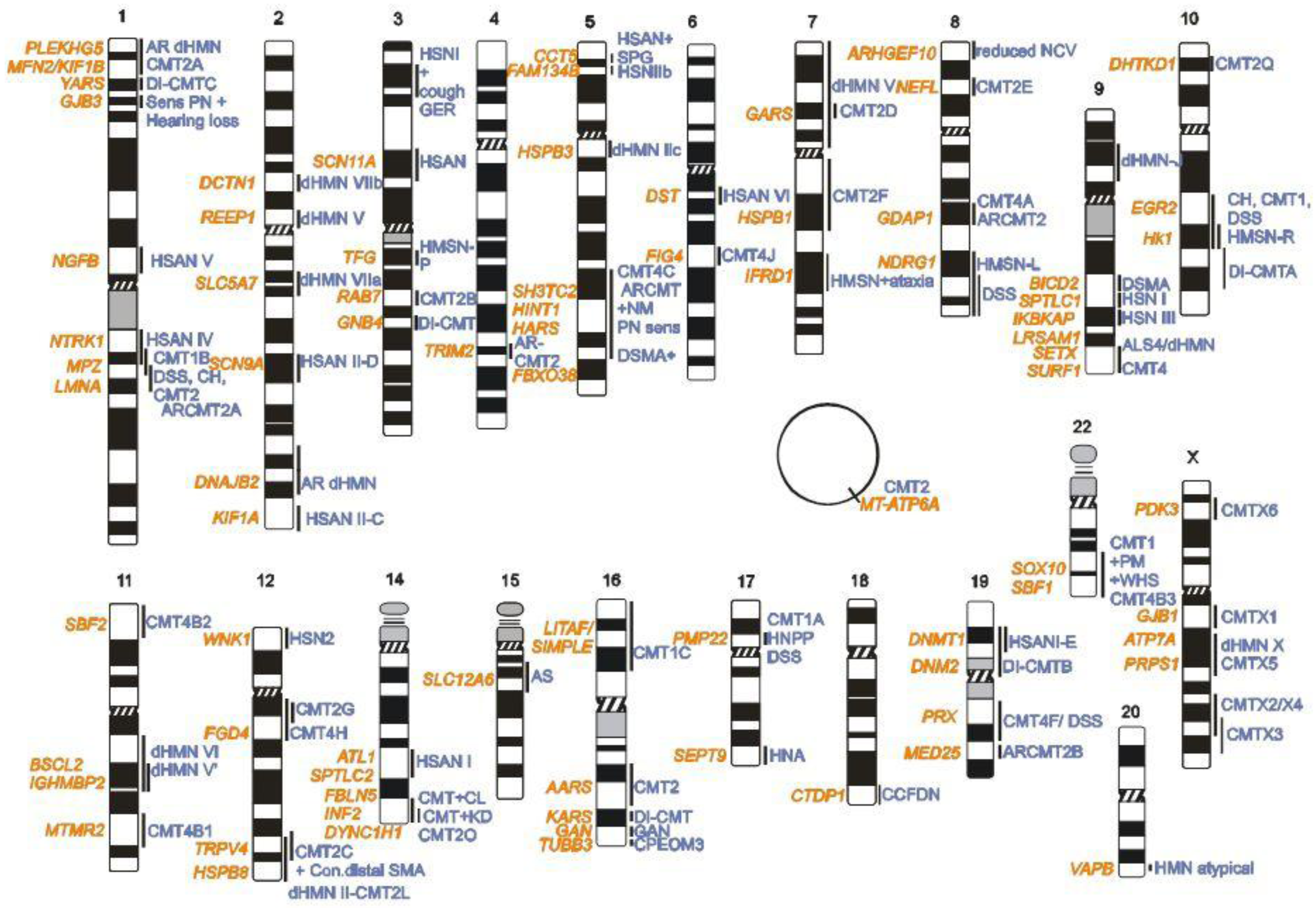
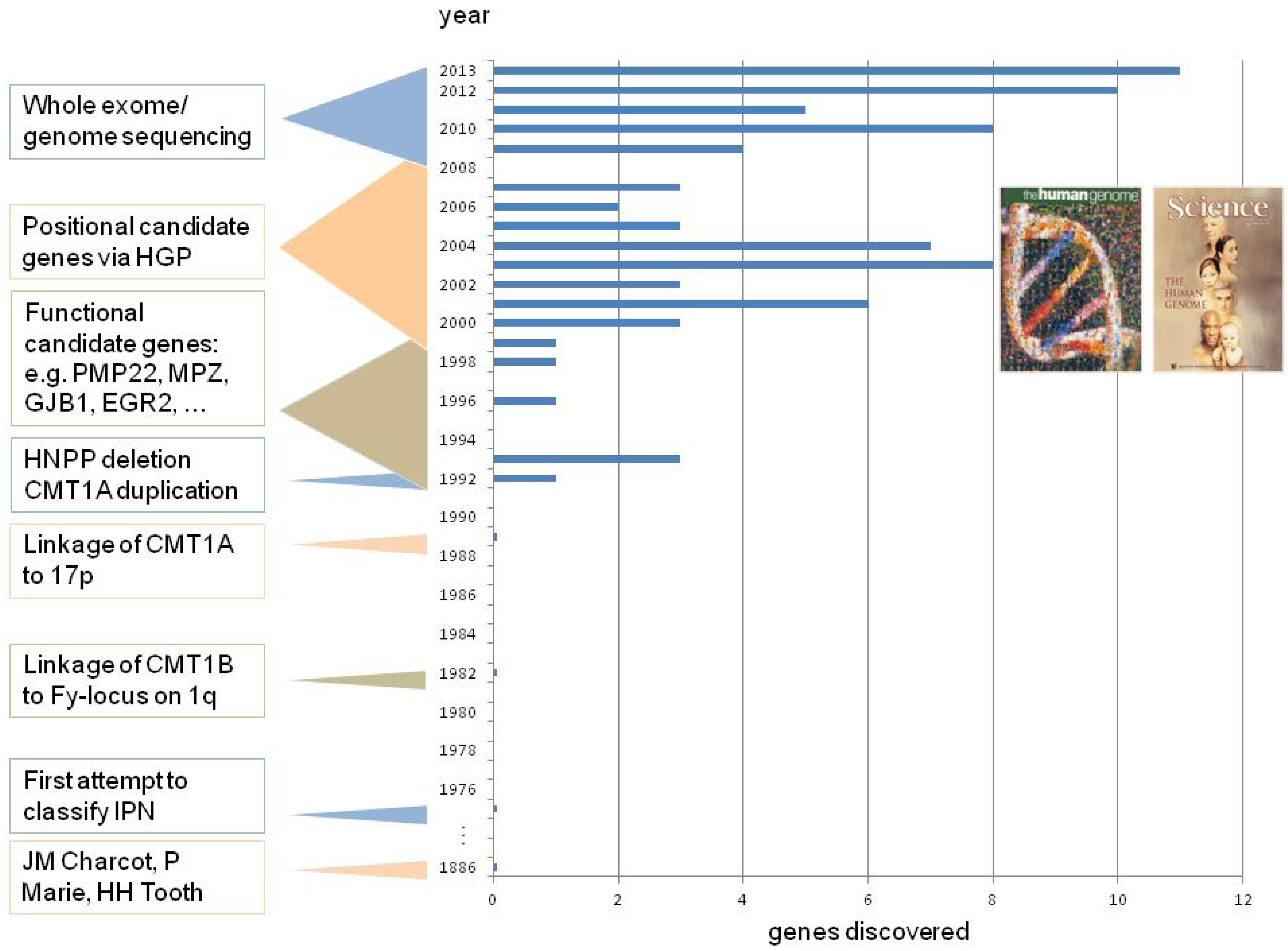
2. CMT Genetics as a Pioneer for Genomic Mechanisms and Emerging Genome Technologies
2.1. Early Linkage Studies
2.2. CMT1A—The First ‘Genomic Disorder’
2.3. Genetic and Physical Mapping, and the Contribution of the Human Genome Reference to Gene Finding in CMT
2.4. CMT2A—The Importance of a Finished Human Genome Reference
3. Next Generation Sequencing Boosted the Identification of CMT Associated Genes
3.1. Targeted Next-Generation Sequencing and Its Limitation in CMT Gene Finding
3.2. Whole Exome Sequencing as a Successful Approach in CMT Gene Finding
3.3. First Whole-Genome Sequencing of a CMT Patient
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Charcot, J.-M.; Marie, P. Sur une forme particulière d’atrophie musculaire progressive, souvent familiale, debutant par les pieds et les jambes et atteignant plus tard les mains. Rev. Med. 1886, 6, 97–138. (in French). [Google Scholar]
- Tooth, H.H. The Peroneal Type of Progressive Muscular Atrophy; H.K. Lewis and Co.: London, UK, 1886. [Google Scholar]
- Dyck, P.J. Definition and basis of classification of hereditary neuropathy with neuronal atrophy and degeneration. In Peripheral Neuropathy, 1st ed.; Dyck, P.J., Thomas, P.K., Lambert, E.H., Eds.; W.B. Saunders Company: Philadelphia, PA, USA, 1975; pp. 825–867. [Google Scholar]
- Dyck, P.J.; Thomas, P.K.; Griffin, J.W.; Low, P.A.; Poduslo, J.F. Peripheral Neuropathy, 4th ed.; WB Saunders: Philadelphia, PA, USA, 2005. [Google Scholar]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef]
- Timmerman, V.; Clowes, V.E.; Reid, E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp. Neurol. 2013, 246, 14–25. [Google Scholar] [CrossRef]
- Reilly, M.M.; Murphy, S.M.; Laura, M. Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 1–14. [Google Scholar] [CrossRef]
- Saporta, M.A.; Shy, M.E. Inherited peripheral neuropathies. Neurol. Clin. 2013, 31, 597–619. [Google Scholar] [CrossRef]
- Bird, T.D.; Ott, J.; Giblett, E.R. Evidence for linkage of Charcot-Marie-Tooth neuropathy to the Duffy locus on chromosome 1. Am. J. Hum. Genet. 1982, 34, 388–394. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Murphy, S.M.; Cottenie, E.; Chalasani, A.; Sweeney, M.G.; Woodward, C.; Mudanohwo, E.E.; Hargreaves, I.; Heales, S.; Land, J.; et al. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology 2012, 79, 1145–1154. [Google Scholar] [CrossRef]
- Suter, U.; Scherer, S.S. Disease mechanisms in inherited neuropathies. Nat. Rev. Neurosci. 2003, 4, 714–726. [Google Scholar] [CrossRef]
- Ingenuity Systems. Available online: http://www.ingenuity.com/ (accessed on 18 October 2013).
- Online Mendelian Inheritance in Man Database (OMIM). Available online: http://ncbi.nlm.nih.gov/omim/ (accessed on 20 November 2013).
- Inherited Peripheral Neuropathy Mutation Database (IPNMDB). Available online: http://molgen.vib-ua.be/CMTMutations/ (accessed on 20 November 2013).
- Leiden Open (Source) Variation Database (LOVD). Available online: http://lovd.nl/ (accessed on 20 November 2013).
- Bird, T.D. Historical perspective of defining Charcot-Marie-Tooth type 1B. Ann. N. Y. Acad. Sci. 1999, 883, 6–13. [Google Scholar] [CrossRef]
- Vance, J.M.; Nicholson, G.A.; Yamaoka, L.H.; Stajich, J.; Stewart, J.S.; Speer, M.C.; Hung, W.-J.; Roses, A.D.; Barker, D.; Pericak-Vance, M.A. Linkage of Charcot-Marie-Tooth neuropathy type 1a to chromosome 17. Exp. Neurol. 1989, 104, 186–189. [Google Scholar] [CrossRef]
- Raeymaekers, P.; Timmerman, V.; de Jonghe, P.; Swerts, L.; Gheuens, J.; Martin, J.-J.; Muylle, L.; de Winter, G.; Vandenberghe, A.; van Broeckhoven, C. Localization of the mutation in an extended family with Charcot- Marie-Tooth neuropathy (HMSN I). Am. J. Hum. Genet. 1989, 45, 953–958. [Google Scholar]
- Middleton-Price, H.R.; Harding, A.E.; Monteiro, C.; Berciano, J.; Malcolm, S. Linkage of hereditary motor and sensory neuropathy type I to the pericentromeric region of chromosome 17. Am. J. Hum. Genet. 1990, 46, 92–94. [Google Scholar]
- Hayasaka, K.; Himoro, M.; Sato, W.; Takada, G.; Uyemura, K.; Shimizu, N.; Bird, T.; Conneally, P.M.; Chance, P.F. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 1993, 5, 31–34. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Guiochon-Mantel, A.; Lacroix, C.; Lapresle, J.; Said, G. The Roussy-Levy family: From the original description to the gene. Ann. Neurol. 1999, 46, 770–773. [Google Scholar] [CrossRef]
- Pareyson, D.; Menichella, D.; Botti, S.; Sghirlanzoni, A.; Fallica, E.; Mora, M.; Ciano, C.; Shy, M.E.; Taroni, F. Heterozygous null mutation in the P0 gene associated with mild Charcot-Marie-Tooth disease. Ann. N. Y. Acad. Sci. 1999, 883, 477–480. [Google Scholar] [CrossRef]
- Warner, L.E.; Hilz, M.J.; Appel, S.H.; Killian, J.M.; Kolodny, E.H.; Karpati, G.; Watters, G.V.; Nelis, E.; van Broeckhoven, C.; Lupski, J.R. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth 1B, Dejerine-Sottas and congenital hypomyelination. Neuron 1996, 17, 451–460. [Google Scholar] [CrossRef]
- Schiavon, F.; Rampazzo, A.; Merlini, L.; Angelini, C.; Mostacciuolo, M.L. Mutations of the same sequence of the myelin P0 gene causing two different phenotypes. Hum. Mutat. 1998, 11, S217–S219. [Google Scholar] [CrossRef]
- De Jonghe, P.; Timmerman, V.; Ceuterick, C.; Nelis, E.; de Vriendt, E.; Löfgren, A.; Vercruyssen, A.; Verellen, C.; van Maldergem, L.; Martin, J.-J.; et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 1999, 122, 281–290. [Google Scholar] [CrossRef]
- Nelis, E.; van Broeckhoven, C.; de Jonghe, P.; Löfgren, A.; Vandenberghe, A.; Latour, P.; Le Guern, E.; Brice, A.; Mostacciuolo, M.L.; Schiavon, F.; et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: A European collaborative study. Eur. J. Hum. Genet. 1996, 4, 25–33. [Google Scholar]
- Szigeti, K.; Garcia, C.; Lupski, J.R. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: Molecular diagnostics determine aspects of medical management. Genet. Med. 2006, 8, 86–92. [Google Scholar] [CrossRef]
- Patel, P.I.; Franco, B.; Garcia, C.; Slaugenhaupt, S.A.; Nakamura, Y.; Ledbetter, D.H.; Chakravarti, A.; Lupski, J.R. Genetic mapping of autosomal dominant Charcot-Marie-Tooth disease in a large French-Acadian kindred: Identification of new linked markers on chromosome 17. Am. J. Hum. Genet. 1990, 46, 801–809. [Google Scholar]
- Timmerman, V.; Raeymaekers, P.; de Jonghe, P.; de Winter, G.; Swerts, L.; Jacobs, K.; Gheuens, J.; Martin, J.-J.; Vandenberghe, A.; van Broeckhoven, C. Assignment of the Charcot-Marie-Tooth neuropathy type 1 (CMT 1a) gene to 17p11.2-p12. Am. J. Hum. Genet. 1990, 47, 680–685. [Google Scholar]
- Raeymaekers, P.; Timmerman, V.; Nelis, E.; de Jonghe, P.; Hoogendijk, J.E.; Baas, F.; Barker, D.F.; Martin, J.-J.; de Visser, M.; Bolhuis, P.A.; et al. HMSN Collaborative Research Group Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscul. Disord. 1991, 1, 93–97. [Google Scholar] [CrossRef]
- Lupski, J.R.; Montes de Oca-Luna, R.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–239. [Google Scholar] [CrossRef]
- Timmerman, V.; Lupski, J.R. The CMT1A duplication and HNPP deletion. In Genomic Disorders: The Genomic Basis of Disease, 1st ed.; Lupski, J.R., Stankiewicz, P., Eds.; Humana Press: Totowa, NJ, USA, 2006. [Google Scholar]
- Chance, P.F.; Alderson, M.K.; Leppig, K.A.; Lensch, M.W.; Matsunami, N.; Smith, B.; Swanson, P.D.; Odelberg, S.J.; Distsche, C.M.; Bird, T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993, 72, 143–151. [Google Scholar] [CrossRef]
- Reiter, L.T.; Murakami, T.; Koeuth, T.; Pentao, L.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat. Genet. 1996, 12, 288–297. [Google Scholar] [CrossRef]
- Kennerson, M.L.; Nassif, N.T.; Dawkins, J.L.; DeKroon, R.M.; Yang, J.G.; Nicholson, G.A. The Charcot-Marie-Tooth binary repeat contains a gene transcribed from the opposite strand of a partially duplicated region of the COX10 gene. Genomics 1997, 46, 61–69. [Google Scholar] [CrossRef]
- Inoue, K.; Dewar, K.; Katsanis, N.; Reiter, L.T.; Lander, E.S.; Devon, K.L.; Wyman, D.W.; Lupski, J.R.; Birren, B. The 1.4 Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res. 2001, 11, 1018–1033. [Google Scholar] [CrossRef]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R.; et al. Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef]
- Patel, P.I.; Roa, B.B.; Welcher, A.A.; Schoener-Scott, R.; Trask, B.J.; Pentao, L.; Snipes, G.J.; Garcia, C.A.; Francke, U.; Shooter, E.M.; et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 159–165. [Google Scholar] [CrossRef]
- Timmerman, V.; Nelis, E.; van Hul, W.; Nieuwenhuijsen, B.W.; Chen, K.L.; Wang, S.; Ben Othman, K.; Cullen, B.; Leach, R.J.; Hanemann, C.O.; et al. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat. Genet. 1992, 1, 171–175. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Bolhuis, P.A.; Zorn, I.; Hoogendijk, J.E.; van den Bosch, N.; Hensels, G.W.; Stanton, V., Jr.; Housman, D.E.; Fischbeck, K.H.; Ross, D.A.; et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot- Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 166–170. [Google Scholar] [CrossRef]
- Lupski, J.R.; Wise, C.A.; Kuwano, A.; Pentao, L.; Parker, J.; Glaze, D.; Ledbetter, D.; Greenberg, F.; Patel, P.I. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 29–33. [Google Scholar] [CrossRef]
- Palau, F.; Löfgren, A.; de Jonghe, P.; Bort, S.; Nelis, E.; Sevilla, T.; Martin, J.-J.; Vílchez, J.; Prieto, F.; van Broeckhoven, C. Origin of the de novo duplication in Charcot-Marie-Tooth disease type 1A: Unequal nonsister chromatid exchange during spermatogenesis. Hum. Mol. Genet. 1993, 2, 2031–2035. [Google Scholar] [CrossRef]
- Zhang, F.; Seeman, P.; Liu, P.; Weterman, M.A.; Gonzaga-Jauregui, C.; Towne, C.F.; Batish, S.D.; de Vriendt, E.; de Jonghe, P.; Rautenstrauss, B.; et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: Rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010, 86, 892–903. [Google Scholar] [CrossRef]
- Boone, P.M.; Wiszniewski, W.; Lupski, J.R. Genomic medicine and neurological disease. Hum. Genet. 2011, 130, 103–121. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef]
- Suter, U.; Welcher, A.A.; Özcelik, T.; Snipes, G.; Kosaras, B.; Francke, U.; Billings-Gagliardi, S. Trembler mouse carries a point mutation in a myelin gene. Nature 1992, 356, 241–244. [Google Scholar] [CrossRef]
- Suter, U.; Moskow, J.J.; Welcher, A.A.; Snipes, G.; Kosaras, B.; Sidman, R.; Buchberg, A.; Shooter, E. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc. Natl. Acad. Sci. USA 1992, 89, 4382–4386. [Google Scholar] [CrossRef]
- Fledrich, R.; Stassart, R.M.; Sereda, M.W. Murine therapeutic models for Charcot-Marie-Tooth (CMT) disease. Br. Med. Bull. 2012, 102, 89–113. [Google Scholar] [CrossRef]
- Sereda, M.W.; zu Horste, G.M.; Suter, U.; Uzma, N.; Nave, K.-A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat. Med. 2003, 9, 1533–1537. [Google Scholar] [CrossRef]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontes, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef]
- Verhamme, C.; de Haan, R.J.; Vermeulen, M.; Baas, F.; de Visser, M.; van Schaik, I.N. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: A randomised, double-blind, placebo-controlled phase II trial. BMC Med. 2009, 7, 70. [Google Scholar] [CrossRef]
- Pareyson, D.; Schenone, A.; Rizzuto, N.; Fabrizi, G.M.; Santoro, L.; Vita, G.; Quattrone, A.; Padua, L.; Gemignani, F.; Visioli, F.; et al. Clinical and electrophysiological evaluation of 222 patients with Charcot-Marie-Tooth disease type 1A recruited in the CMT-TRIAAL (ascorbic acid therapy for Charcot-Marie-Tooth 1A disease). J. Neurol. 2008, 255, 104–105. [Google Scholar]
- Pareyson, D.; Schenone, A.; Fabrizi, G.M.; Santoro, L.; Padua, L.; Quattrone, A.; Vita, G.; Gemignani, F.; Visioli, F.; Solari, A.; et al. A multicenter, randomized, double-blind, placebo-controlled trial of long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL): The study protocol [EudraCT no.: 2006-000032-27]. Pharmacol. Res. 2006, 54, 436–441. [Google Scholar] [CrossRef]
- Burns, J.; Ouvrier, R.A.; Yiu, E.M.; Joseph, P.D.; Kornberg, A.J.; Fahey, M.C.; Ryan, M.M. Ascorbic acid for Charcot-Marie-Tooth disease type 1A in children: A randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol. 2009, 8, 537–544. [Google Scholar] [CrossRef]
- Lewis, R.A.; McDermott, M.P.; Herrmann, D.N.; Hoke, A.; Clawson, L.L.; Siskind, C.; Feely, S.M.; Miller, L.J.; Barohn, R.J.; Smith, P.; et al. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: Results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013, 70, 981–987. [Google Scholar] [CrossRef]
- Micallef, J.; Attarian, S.; Dubourg, O.; Gonnaud, P.M.; Hogrel, J.Y.; Stojkovic, T.; Bernard, R.; Jouve, E.; Pitel, S.; Vacherot, F.; et al. Effect of ascorbic acid in patients with Charcot-Marie-Tooth disease type 1A: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2009, 8, 1103–1110. [Google Scholar] [CrossRef]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Aretz, S.; Rautenstrauss, B.; Timmerman, V. Clinical utility gene card for: HMSN/HNPP HMSN types 1, 2, 3, 6 (CMT1,2,4, DSN, CHN, GAN, CCFDN, HNA); HNPP. Eur. J. Hum. Genet. 2010, 18. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. Hereditary distal spinal muscular atrophy. A report on 34 cases and a review of the literature. J. Neurol. Sci. 1980, 45, 337–348. [Google Scholar] [CrossRef]
- Irobi, J.; Dierick, I.; Jordanova, A.; Claeys, K.; de Jonghe, P.; Timmerman, V. Unravelling the genetics of distal hereditary motor neuronopathies. NeuroMol. Med. 2006, 8, 131–146. [Google Scholar] [CrossRef]
- Timmerman, V.; Raeymaekers, P.; Nelis, E.; de Jonghe, P.; Muylle, L.; Ceuterick, C.; Martin, J.-J.; van Broeckhoven, C. Linkage analysis of distal hereditary motor neuropathy type II (distal HMN II) in a single pedigree. J. Neurol. Sci. 1992, 109, 41–48. [Google Scholar] [CrossRef]
- Gyapay, G.; Morissette, J.; Vignal, A.; Dib, C.; Fizames, C.; Millasseau, P.; Marc, S.; Bernardi, G.; Lathrop, M.; Weissenbach, J. The 1993–94 Genethon human genetic linkage map [see comments]. Nat. Genet. 1994, 7, 246–339. [Google Scholar] [CrossRef]
- Timmerman, V.; de Jonghe, P.; Simokovic, S.; Löfgren, A.; Beuten, J.; Nelis, E.; Ceuterick, C.; Martin, J.-J.; van Broeckhoven, C. Distal hereditary motor neuropathy type II (distal HMN II): Mapping of a locus to chromosome 12q24. Hum. Mol. Genet. 1996, 5, 1065–1069. [Google Scholar] [CrossRef]
- Irobi, J.; Tissir, F.; de Jonghe, P.; de Vriendt, E.; van Broeckhoven, C.; Timmerman, V.; Beuten, J. A clone contig of 12q24.3 encompassing the distal hereditary motor neuropathy type II gene. Genomics 2000, 65, 34–43. [Google Scholar] [CrossRef]
- Irobi, J.; van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; de Vriendt, E.; Jacobs, A.; van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Ismailov, S.M.; Fedotov, V.P.; Dadali, E.L.; Polyakov, A.V.; van Broeckhoven, C.; Ivanov, V.I.; de Jonghe, P.; Timmerman, V.; Evgrafov, O.V. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2F) maps to chromosome 7q11-q21. Eur. J. Hum. Genet. 2001, 9, 646–650. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.V.; Irobi, J.; van den Bosch, L.; Dierick, I.; Schagina, O.; Verpoorten, N.; van Impe, K.; Fedotov, V.P.; Dadali, E.L.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef]
- Holmgren, A.; Bouhy, D.; Timmerman, V. Molecular Biology of small HSPs associated with Peripheral Neuropathies. eLS 2012. [Google Scholar] [CrossRef]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; van den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Saito, M.; Hayashi, Y.; Suzuki, T.; Tanaka, H.; Hozumi, I.; Tsuji, S. Linkage mapping of the gene for Charcot-Marie-Tooth disease type 2 to chromosome 1p (CMT2A) and the clinical features of CMT2A. Neurology 1997, 49, 1630–1635. [Google Scholar] [CrossRef]
- Zhao, C.; Takita, J.; Tanaka, Y.; Setou, M.; Nakagawa, T.; Takeda, S.; Wei Yang, H.; Terada, S.; Nakata, T.; Takei, Y.; et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 2001, 105, 587–597. [Google Scholar] [CrossRef]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Verhoeven, K.; Claeys, K.; Züchner, S.; Schröder, J.M.; Weis, J.; Ceuterick, C.; Jordanova, A.; Nelis, E.; de Vriendt, E.; van Hul, M.; et al. Mitofusin 2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 2006, 129, 2093–2102. [Google Scholar] [CrossRef]
- Lv, H.; Wang, L.; Li, W.; Qiao, X.; Li, Y.; Wang, Z.; Yuan, Y. Mitofusin 2 gene mutation causing early-onset CMT2A with different progressive courses. Clin. Neuropathol. 2013, 32, 16–23. [Google Scholar] [CrossRef]
- Chung, K.W.; Kim, S.B.; Park, K.D.; Choi, K.G.; Lee, J.H.; Eun, H.W.; Suh, J.S.; Hwang, J.H.; Kim, W.K.; Seo, B.C.; et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain 2006, 129, 2103–2118. [Google Scholar] [CrossRef]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef]
- Züchner, S.; de Jonghe, P.; Jordanova, A.; Claeys, K.; Guergelcheva, V.; Cherninkova, S.; Hamilton, S.R.; van Stavern, G.; Krajewski, K.; Stajich, J.; et al. Axonal neuropathy with optic atrophy (HMSN VI) is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef]
- Zhu, D.; Kennerson, M.L.; Walizada, G.; Züchner, S.; Vance, J.M.; Nicholson, G.A. Charcot-Marie-Tooth with pyramidal signs is genetically heterogeneous: Families with and without MFN2 mutations. Neurology 2005, 65, 496–497. [Google Scholar] [CrossRef]
- McCorquodale, D.S.; Montenegro, G.; Peguero, A.; Carlson, N.; Speziani, F.; Price, J.; Taylor, S.W.; Melanson, M.; Vance, J.M.; Zuchner, S. Mutation screening of mitofusin 2 in Charcot-Marie-Tooth disease type 2. J. Neurol. 2011, 258, 1234–1239. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Weger, M.; Fink-Puches, R.; Papic, L.; Frohlich, E.; Auer-Grumbach, P.; El Shabrawi-Caelen, L.; Schabhuttl, M.; Windpassinger, C.; Senderek, J.; et al. Fibulin-5 mutations link inherited neuropathies, age-related macular degeneration and hyperelastic skin. Brain 2011, 134, 1839–1852. [Google Scholar] [CrossRef]
- Zimon, M.; Baets, J.; Almeida-Souza, L.; de Vriendt, E.; Nikodinovic, J.; Parman, Y.; Battalo Gcaron, L.E.; Matur, Z.; Guergueltcheva, V.; Tournev, I.; et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat. Genet. 2012, 44, 1080–1083. [Google Scholar] [CrossRef]
- Azzedine, H.; Senderek, J.; Rivolta, C.; Chrast, R. Molecular genetics of charcot-marie-tooth disease: From genes to genomes. Mol. Syndromol. 2012, 3, 204–214. [Google Scholar]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef]
- Montenegro, G.; Powell, E.; Huang, J.; Speziani, F.; Edwards, Y.J.; Beecham, G.; Hulme, W.; Siskind, C.; Vance, J.; Shy, M.; et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann. Neurol. 2011, 69, 464–470. [Google Scholar] [CrossRef]
- Gonzales, M.A.; McLaughlin, H.M.; Houlden, H.; Guo, M. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1247–1249. [Google Scholar] [CrossRef]
- Peeters, K.; Litvinenko, I.; Asselbergh, B.; Almeida-Souza, L.; Chamova, T.; Geuens, T.; Ydens, E.; Zimon, M.; Irobi, J.; de Vriendt, E.; et al. Molecular defects in the motor adaptor BICD2 Cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am. J. Hum. Genet. 2013, 92, 955–964. [Google Scholar] [CrossRef]
- Neveling, K.; Martinez-Carrera, L.A.; Holker, I.; Heister, A.; Verrips, A.; Hosseini-Barkooie, S.M.; Gilissen, C.; Vermeer, S.; Pennings, M.; Meijer, R.; et al. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am. J. Hum. Genet. 2013, 92, 946–954. [Google Scholar] [CrossRef]
- Oates, E.C.; Rossor, A.M.; Hafezparast, M.; Gonzalez, M.; Speziani, F.; Macarthur, D.G.; Lek, M.; Cottenie, E.; Scoto, M.; Foley, A.R.; et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 965–973. [Google Scholar] [CrossRef]
- Kennerson, M.L.; Yiu, E.M.; Chuang, D.T.; Kidambi, A.; Tso, S.C.; Ly, C.; Chaudhry, R.; Drew, A.P.; Rance, G.; Delatycki, M.B.; et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum. Mol. Genet. 2013, 22, 1404–1416. [Google Scholar] [CrossRef]
- Leipold, E.; Liebmann, L.; Korenke, G.C.; Heinrich, T.; Giesselmann, S.; Baets, J.; Ebbinghaus, M.; Goral, R.O.; Stodberg, T.; Hennings, J.C.; et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 2013, 45, 1399–1404. [Google Scholar] [CrossRef]
- Barwick, K.E.; Wright, J.; Al-Turki, S.; McEntagart, M.M.; Nair, A.; Chioza, B.; Al-Memar, A.; Modarres, H.; Reilly, M.M.; Dick, K.J.; et al. Defective presynaptic choline transport underlies hereditary motor neuropathy. Am. J. Hum. Genet. 2012, 91, 1103–1107. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; van Maldergem, L.; He, W.; Chan, W.M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio, D.D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A.; et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef]
- Züchner, S. Peripheral neuropathies: Whole genome sequencing identifies causal variants in CMT. Nat. Rev. Neurol. 2010, 6, 424–425. [Google Scholar] [CrossRef]
- Senderek, J.; Bergmann, C.; Stendel, C.; Kirfel, J.; Verpoorten, N.; de Jonghe, P.; Timmerman, V.; Chrast, R.; Verheijen, M.H.G.; Lemke, G.; et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 2003, 73, 1106–1119. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Lebrigio, R.F.; van Booven, D.; Ulloa, R.H.; Powell, E.; Speziani, F.; Tekin, M.; Schule, R.; Zuchner, S. GEnomes Management Application (GEM.app): A new software tool for large-scale collaborative genome analysis. Hum. Mutat. 2013, 34, 842–846. [Google Scholar] [CrossRef]
- Genome Variant Database for Human Diseases. Available online: http://www.genomics.med.miami.edu/ (accessed on 20 November 2013).
- Human Gene Mutation Database. Available online: http://www.biobase-international.com/product/hgmd/ (accessed on 20 November 2013).
- Inherited Neuropathy Consortium. Available online: http://rarediseasesnetwork.epi.usf.edu/INC/ (accessed on 20 November 2013).
- Niemann, A.; Berger, P.; Suter, U. Pathornechanisms of mutant proteins in Charcot-Marie-Tooth disease. NeuroMol. Med. 2006, 8, 217–241. [Google Scholar] [CrossRef]
- Bouhy, D.; Timmerman, V. Animal models and therapeutic prospects for Charcot-Marie-Tooth disease. Ann. Neurol. 2013, 74, 391–396. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13-32. https://doi.org/10.3390/genes5010013
Timmerman V, Strickland AV, Züchner S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes. 2014; 5(1):13-32. https://doi.org/10.3390/genes5010013
Chicago/Turabian StyleTimmerman, Vincent, Alleene V. Strickland, and Stephan Züchner. 2014. "Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success" Genes 5, no. 1: 13-32. https://doi.org/10.3390/genes5010013
APA StyleTimmerman, V., Strickland, A. V., & Züchner, S. (2014). Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes, 5(1), 13-32. https://doi.org/10.3390/genes5010013