Copy Number Variation in Hereditary Non-Polyposis Colorectal Cancer

 ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Samples
2.2. Genomic Array Analysis
2.3. Statistical, Pathway and Annotation Analysis
3. Results and Discussion
3.1. Resolution
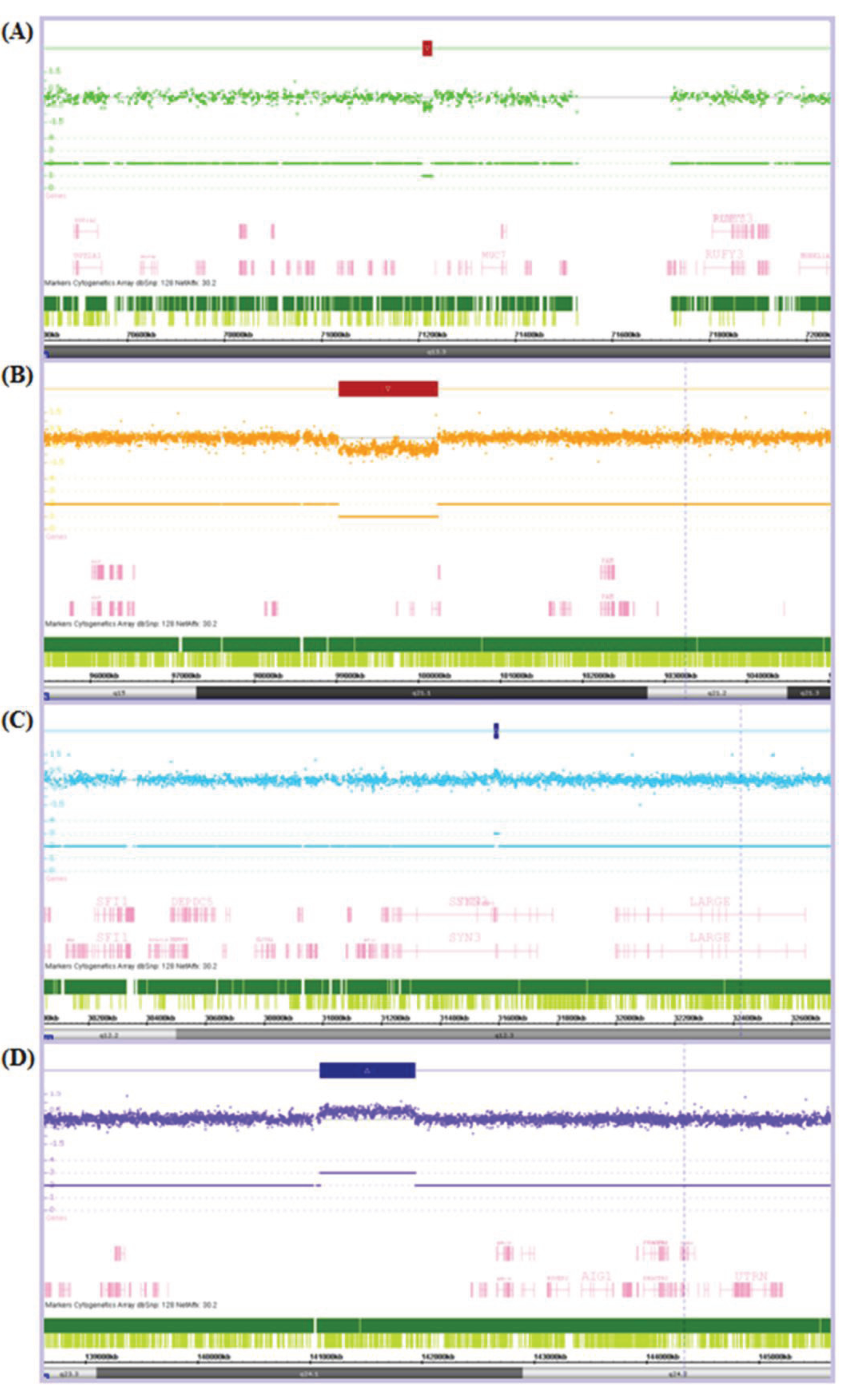
3.2. CNV Detection

{kind=link}
| CNV Count | CNV Size (Kb) | ||||||
|---|---|---|---|---|---|---|---|
| Participants | Total CNVs per group | Median CNVs per sample | Mean CNVs per sample | Total CNV affected genome per group | Mean total CNV affected genome per sample | Mean size of a CNV | |
| Patients | 125 | 439 | 2 | 3.51 | 35,508.53 | 284.07 | 70.08 |
| Controls | 40 | 104 | 2 | 2.60 | 11,820.75 | 295.52 | 106.57 |
| p | − | − | − | 0.2980 | − | 0.9121 | 0.0165 * |
3.3. MMR Gene Interrogation
| Gene | Chr. | Start (bp) | End (bp) | Size (Kb) | Search start (bp) | Search end (bp) |
|---|---|---|---|---|---|---|
| EXO1 | 1 | 240,078,157 | 240,119,671 | 42 | 240,028,157 | 240,169,671 |
| RPA2 | 1 | 28,090,635 | 28,113,823 | 23 | 28,040,635 | 28,163,823 |
| MSH2 | 2 | 47,783,766 | 47,563,864 | 80 | 47,733,766 | 47,613,864 |
| MSH6 | 2 | 47,863,724 | 47,887,596 | 24 | 47,813,724 | 47,937,596 |
| PMS1 | 2 | 190,357,055 | 190,450,600 | 94 | 190,307,055 | 190,500,600 |
| MLH1 | 3 | 37,009,982 | 37,067,341 | 57 | 36,959,982 | 37,117,341 |
| RFC4 | 3 | 187,990,375 | 188,007,178 | 17 | 187,940,375 | 188,057,178 |
| RFC1 | 4 | 38,965,470 | 39,044,390 | 79 | 38,915,470 | 39,094,390 |
| MSH3 | 5 | 79,986,049 | 80,208,390 | 222 | 79,936,049 | 80,258,390 |
| PMS2 | 7 | 5,979,395 | 6,015,263 | 36 | 5,929,395 | 6,065,263 |
| POLD2 | 7 | 44,120,810 | 44,129,672 | 9 | 44,070,810 | 44,179,672 |
| RFC2 | 7 | 73,283,767 | 73,306,674 | 23 | 73,233,767 | 73,356,674 |
| RPA3 | 7 | 7,643,099 | 7,724,763 | 82 | 7,593,099 | 7,774,763 |
| POLD3 | 11 | 73,981,276 | 74,031,413 | 50 | 73,931,276 | 74,081,413 |
| POLD4 | 11 | 66,875,594 | 66,877,593 | 2 | 66,825,594 | 66,927,593 |
| RFC5 | 12 | 116,938,890 | 116,954,422 | 16 | 116,888,890 | 117,004,422 |
| RFC3 | 13 | 33,290,205 | 33,438,695 | 148 | 33,240,205 | 33,488,695 |
| MLH3 | 14 | 74,550,219 | 74,587,988 | 38 | 74,500,219 | 74,637,988 |
| RPA1 | 17 | 1,680,022 | 1,749,598 | 70 | 1,630,022 | 1,799,598 |
| LIG1 | 19 | 53,310,514 | 53,365,372 | 55 | 53,260,514 | 53,415,372 |
| POLD1 | 19 | 55,579,404 | 55,613,083 | 34 | 55,529,404 | 55,663,083 |
| PCNA | 20 | 5,043,598 | 5,055,268 | 12 | 4,993,598 | 5,105,268 |
3.4. Occurrence and Distribution of CNVs in Patients and Controls
| Type | 2 CNV events | 3 CNV events | 4 CNV events | |||
|---|---|---|---|---|---|---|
| Gains | ADARB2 | DEFB125 | ITGA1 | PSG8 | GK5 | LOC642597 |
| APC | DEFB126 | KIAA1680 | RBCK1 | IGSF11 | ||
| ARHGAP19 | DEFB127 | LATS2 | RNF125 | LCP1 | ||
| B2M | DEFB128 | MLL | RNF138 | XRN1 | ||
| BBOX1 | DEFB129 | MSI2 | SOX12 | NAMPT | ||
| C10orf139 | DEFB132 | NRSN2 | TBC1D20 | |||
| C14orf23 | EPHA7 | NXPH1 | TFG | |||
| C20orf96 | FAM134B | ODZ4 | TRIB3 | |||
| C3orf33 | FOXG1 | PELO | TRIM69 | |||
| CNTN5 | GPR128 | PHC3 | WDR37 | |||
| CNTNAP2 | GPR160 | PRKCI | ZCCHC3 | |||
| CSNK2A1 | GYPE | PSG10 | ZMYND11 | |||
| Losses | CNTN4 | CTNNA3 | ||||
| DCDC1 | ||||||
| PPP2R3C | ||||||
| Both | NRG3 |
| Chr. | Start (bp) | End (bp) | Size (Kb) | Conf | Probes |
|---|---|---|---|---|---|
| 2 CNV gains | |||||
| 3 | 189,058,439 | 189,098,718 | 40.28 | 0.93 | 31 |
| 3 | 189,069,317 | 189,088,009 | 18.69 | 0.94 | 26 |
| 4 | 44,664,798 | 44,699,744 | 34.95 | 0.92 | 41 |
| 4 | 44,664,798 | 44,699,744 | 34.95 | 0.90 | 41 |
| 8 | 120,414,388 | 120,438,172 | 23.78 | 0.91 | 27 |
| 8 | 120,419,721 | 120,451,773 | 32.05 | 0.91 | 30 |
| 11 | 29,547,229 | 29,593,722 | 46.49 | 0.90 | 39 |
| 11 | 29,547,756 | 29,593,722 | 45.97 | 0.91 | 37 |
| 16 | 25,330,672 | 25,438,375 | 107.70 | 0.92 | 46 |
| 16 | 25,330,672 | 25,438,375 | 107.70 | 0.92 | 46 |
| 3 CNV gains | |||||
| 3 | 19,014,033 | 19,041,376 | 27.34 | 0.90 | 31 |
| 3 | 19,016,875 | 19,041,376 | 24.50 | 0.91 | 28 |
| 3 | 19,016,875 | 19,041,376 | 24.50 | 0.91 | 28 |
| 5 | 59,744,695 | 59,807,906 | 63.21 | 0.92 | 52 |
| 5 | 59,744,695 | 59,811,770 | 67.08 | 0.93 | 54 |
| 5 | 59,749,693 | 59,807,906 | 58.21 | 0.92 | 51 |
| 9 | 103,982,826 | 104,016,588 | 33.76 | 0.91 | 27 |
| 9 | 103,982,826 | 104,017,715 | 34.89 | 0.90 | 28 |
| 9 | 103,991,205 | 104,017,715 | 26.51 | 0.91 | 26 |
| 11 | 15,765,333 | 15,791,331 | 26.00 | 0.90 | 30 |
| 11 | 15,770,233 | 15,796,302 | 26.07 | 0.92 | 30 |
| 11 | 15,776,946 | 15,795,665 | 18.72 | 0.91 | 24 |
| 12 | 16,469,855 | 16,503,960 | 34.11 | 0.91 | 33 |
| 12 | 16,469,855 | 16,503,960 | 34.11 | 0.91 | 33 |
| 12 | 16,476,470 | 16,506,851 | 30.38 | 0.92 | 33 |
| 4 CNV gains | |||||
| 16 | 63,364,955 | 63,389,659 | 24.70 | 0.91 | 33 |
| 16 | 63,369,029 | 63,389,029 | 20.00 | 0.92 | 30 |
| 16 | 63,369,960 | 63,388,189 | 18.23 | 0.90 | 28 |
| 16 | 63,371,038 | 63,397,352 | 26.31 | 0.92 | 38 |
| 5 CNV gains | |||||
| 5 | 116,651,923 | 116,698,621 | 46.70 | 0.91 | 36 |
| 5 | 116,655,439 | 116,692,153 | 36.71 | 0.92 | 27 |
| 5 | 116,656,039 | 116,695,730 | 39.69 | 0.91 | 29 |
| 5 | 116,660,694 | 116,697,347 | 36.65 | 0.93 | 28 |
| 5 | 116,660,694 | 116,693,035 | 32.34 | 0.91 | 24 |
| 2 CNV losses | |||||
| 1 | 82,801,000 | 82,821,932 | 20.93 | 0.93 | 31 |
| 1 | 82,801,000 | 82,821,932 | 20.93 | 0.94 | 31 |
| 2 | 22,087,558 | 22,261,901 | 174.34 | 0.91 | 110 |
| 2 | 22,087,558 | 22,261,901 | 174.34 | 0.93 | 110 |
| 2 | 215,167,158 | 215,204,595 | 37.44 | 0.93 | 49 |
| 2 | 215,167,158 | 215,204,595 | 37.44 | 0.91 | 49 |
| 3 | 6,562,398 | 6,603,706 | 41.31 | 0.94 | 42 |
| 3 | 6,562,398 | 6,603,706 | 41.31 | 0.93 | 42 |
| 3 | 166,523,809 | 166,565,186 | 41.38 | 0.95 | 39 |
| 3 | 166,525,250 | 166,565,186 | 39.94 | 0.93 | 38 |
| 5 | 61,460,851 | 61,504,678 | 43.83 | 0.95 | 31 |
| 5 | 61,460,851 | 61,504,678 | 43.83 | 0.93 | 31 |
| 7 | 92,319,307 | 92,343,906 | 24.60 | 0.94 | 26 |
| 7 | 92,319,307 | 92,343,906 | 24.60 | 0.94 | 26 |
| 9 | 104,331,902 | 104,396,632 | 64.73 | 0.96 | 35 |
| 9 | 104,331,902 | 104,396,632 | 64.73 | 0.96 | 35 |
| 5 CNV losses | |||||
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.93 | 26 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.94 | 26 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.96 | 26 |
| 3 | 177,370,126 | 177,399,625 | 29.50 | 0.93 | 27 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.93 | 26 |
| 2 CNV gain and loss | |||||
| 7 * | 110,748,452 | 111,047,157 | 298.71 | 0.93 | 291 |
| 7 ** | 111,007,466 | 111,052,498 | 45.03 | 0.94 | 25 |
| 3 ** | 21,228,980 | 21,313,310 | 84.33 | 0.90 | 88 |
| 3 * | 21,273,619 | 21,339,035 | 65.42 | 0.92 | 62 |
3.5. Pathway Analysis
| KEGG Pathway | Genes in Pathway | Observed | Expected | p |
|---|---|---|---|---|
| Carbohydrate digestion and absorption | 44 | 5 | 0.32 | 0.0012 |
| Starch and sucrose metabolism | 54 | 5 | 0.4 | 0.0017 |
| Metabolic pathways | 1,130 | 21 | 8.31 | 0.0023 |
| Salivary secretion | 89 | 5 | 0.65 | 0.0058 |
| Tight junction | 132 | 6 | 0.97 | 0.0058 |
| Neurotrophin signaling pathway | 127 | 6 | 0.93 | 0.0058 |
| Propanoate metabolism | 32 | 3 | 0.24 | 0.017 |
| Valine, leucine and isoleucine degradation | 44 | 3 | 0.32 | 0.0251 |
| Prostate cancer | 89 | 4 | 0.65 | 0.0251 |
| Ribosome biogenesis in eukaryotes | 80 | 4 | 0.59 | 0.0251 |
| ErbB signaling pathway | 87 | 4 | 0.64 | 0.0251 |
| mRNA surveillance pathway | 83 | 4 | 0.61 | 0.0251 |
| Terpenoid backbone biosynthesis | 15 | 2 | 0.11 | 0.0285 |
| Malaria | 51 | 3 | 0.37 | 0.0293 |
| Endometrial cancer | 52 | 3 | 0.38 | 0.0293 |
| Glycerolipid metabolism | 50 | 3 | 0.37 | 0.0293 |
| Olfactory transduction | 388 | 8 | 2.85 | 0.0346 |
| beta-Alanine metabolism | 22 | 2 | 0.16 | 0.0439 |
3.6. MicroRNA Annotation
| Category | # sub-categories | # of Sig. Categories | Significant Findings |
|---|---|---|---|
| Family | 22 | 1 | miR-17 |
| Cluster | 33 | 0 | − |
| Function | 39 | 8 | Onco-miRs, Apoptosis, Cell cycle related |
| HMDD | 162 | 20 | Cancer (80%), Cardiovascular (4%), Infection (4%) and Psychological disorders (10%) |
| Tissue | 5 | 1 | Placenta |
3.7. CNV Burden
3.8. CNV Bias
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International collaborative group on hereditary non-polyposis colorectal cancer (icg-hnpcc). Dis. Colon. Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef]
- Kemp, Z.; Thirlwell, C.; Sieber, O.; Silver, A.; Tomlinson, I. An update on the genetics of colorectal cancer. Hum. Mol. Genet. 2004, 13, R177–R185. [Google Scholar] [CrossRef]
- Peltomaki, P. Deficient DNA mismatch repair: A common etiologic factor for colon cancer. Hum. Mol. Genet. 2001, 10, 735–740. [Google Scholar] [CrossRef]
- Thompson, E.; Meldrum, C.J.; Crooks, R.; McPhillips, M.; Thomas, L.; Spigelman, A.D.; Scott, R.J. Hereditary non-polyposis colorectal cancer and the role of hPMS2 and hEXO1 mutations. Clin. Genet. 2004, 65, 215–225. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Vissers, L.E.; Venkatachalam, R.; Bodmer, D.; Hoenselaar, E.; Goossens, M.; Haufe, A.; Kamping, E.; Niessen, R.C.; Hogervorst, F.B.; et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011, 32, 407–414. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- McPhillips, M.; Meldrum, C.J.; Creegan, R.; Edkins, E.; Scott, R.J. Deletion Mutations in an Australian Series of HNPCC Patients. Hered. Cancer Clin. Pract. 2005, 3, 43–47. [Google Scholar] [CrossRef]
- Bonis, P.A.; Trikalinos, T.A.; Chung, M.; Chew, P.; Ip, S.; DeVine, D.A.; Lau, J. Hereditary nonpolyposis colorectal cancer: Diagnostic strategies and their implications. Evid. Rep. Technol. Assess. 2007, 150, 1–180. [Google Scholar]
- Obermair, A.; Youlden, D.R.; Young, J.P.; Lindor, N.M.; Baron, J.A.; Newcomb, P.A.; Parry, S.; Hopper, J.L.; Haile, R.; Jenkins, M.A. Risk of Endometrial Cancer for women diagnosed with HNPCC-related colorectal cancer. Int. J. Cancer 2010, 127, 7. [Google Scholar]
- Ionita-Laza, I.; Rogers, A.J.; Lange, C.; Raby, B.A.; Lee, C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 2009, 93, 22–26. [Google Scholar] [CrossRef]
- Almal, S.H.; Padh, H. Implications of gene copy-number variation in health and diseases. J. Hum. Genet. 2012, 57, 6–13. [Google Scholar] [CrossRef]
- Bronstad, I.; Wolff, A.S.; Lovas, K.; Knappskog, P.M.; Husebye, E.S. Genome-wide copy number variation (CNV) in patients with autoimmune Addison’s disease. BMC Med. Genet. 2011, 12, 111. [Google Scholar] [CrossRef]
- Grozeva, D.; Kirov, G.; Ivanov, D.; Jones, I.R.; Jones, L.; Green, E.K.; St Clair, D.M.; Young, A.H.; Ferrier, N.; Farmer, A.E.; et al. Rare copy number variants: A point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch. Gen. Psychiatry 2010, 67, 318–327. [Google Scholar] [CrossRef]
- Hai, R.; Pei, Y.F.; Shen, H.; Zhang, L.; Liu, X.G.; Lin, Y.; Ran, S.; Pan, F.; Tan, L.J.; Lei, S.F.; et al. Genome-wide association study of copy number variation identified gremlin1 as a candidate gene for lean body mass. J. Hum. Genet. 2012, 57, 33–37. [Google Scholar] [CrossRef]
- Jiang, Q.; Ho, Y.Y.; Hao, L.; Nichols Berrios, C.; Chakravarti, A. Copy number variants in candidate genes are genetic modifiers of Hirschsprung disease. PLoS One 2011, 6, e21219. [Google Scholar]
- Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010, 464, 713–720. [Google Scholar] [CrossRef]
- Delnatte, C.; Sanlaville, D.; Mougenot, J.F.; Vermeesch, J.R.; Houdayer, C.; Blois, M.C.; Genevieve, D.; Goulet, O.; Fryns, J.P.; Jaubert, F.; et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am. J. Hum. Genet. 2006, 78, 1066–1074. [Google Scholar] [CrossRef]
- Van Hattem, W.A.; Brosens, L.A.; de Leng, W.W.; Morsink, F.H.; Lens, S.; Carvalho, R.; Giardiello, F.M.; Offerhaus, G.J. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut 2008, 57, 623–627. [Google Scholar] [CrossRef]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Nagasaka, T.; Rhees, J.; Kloor, M.; Gebert, J.; Naomoto, Y.; Boland, C.R.; Goel, A. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010, 70, 3098–3108. [Google Scholar] [CrossRef]
- McEvoy, M.; Smith, W.; D’Este, C.; Duke, J.; Peel, R.; Schofield, P.; Scott, R.; Byles, J.; Henry, D.; Ewald, B.; et al. Cohort profile: The hunter community study. Int. J. Epidemiol. 2010, 39, 1452–1463. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A national cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: Meeting highlights and bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef]
- GraphPad Software. Available online: http://www.graphpad.com/quickcalcs/ttest1/ (accessed on 27 February 2013).
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Lu, M.; Shi, B.; Wang, J.; Cao, Q.; Cui, Q. TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinf. 2010, 11, 419. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Maner, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef]
- Krepischi, A.C.; Achatz, M.I.; Santos, E.M.; Costa, S.S.; Lisboa, B.C.; Brentani, H.; Santos, T.M.; Goncalves, A.; Nobrega, A.F.; Pearson, P.L.; et al. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012, 14, R24. [Google Scholar] [CrossRef]
- Mitchell, R.J.; Farrington, S.M.; Dunlop, M.G.; Campbell, H. Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: A HuGE review. Am. J. Epidemiol. 2002, 156, 885–902. [Google Scholar] [CrossRef]
- Foran, E.; McWilliam, P.; Kelleher, D.; Croke, D.T.; Long, A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int. J. Cancer 2006, 118, 2098–2104. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, M.; Yoshikawa, T.; Chen, H.; Brown, E.J.; Masuho, Y.; Omata, M.; Seki, N. Differential expression of the L-plastin gene in human colorectal cancer progression and metastasis. Biochem. Biophys. Res. Commun. 2001, 289, 876–881. [Google Scholar] [CrossRef]
- Park, T.; Chen, Z.P.; Leavitt, J. Activation of the leukocyte plastin gene occurs in most human cancer cells. Cancer Res. 1994, 54, 1775–1781. [Google Scholar]
- Sygut, A.; Przybylowska, K.; Ferenc, T.; Dziki, L.; Spychalski, M.; Mik, M.; Dziki, A. Genetic Variations of the CTNNA1 And The CTNNB1 Genes in Sporadic Colorectal Cancer in Polish Population. Pol. Przegl. Chir. 2012, 84, 560–564. [Google Scholar]
- Giannini, A.L.; Vivanco, M.; Kypta, R.M. Alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J. Biol. Chem. 2000, 275, 21883–21888. [Google Scholar] [CrossRef]
- Patel, S.G.; Ahnen, D.J. Familial colon cancer syndromes: An update of a rapidly evolving field. Curr. Gastroenterol. Rep. 2012, 14, 428–438. [Google Scholar] [CrossRef]
- Albuquerque, C.; Baltazar, C.; Filipe, B.; Penha, F.; Pereira, T.; Smits, R.; Cravo, M.; Lage, P.; Fidalgo, P.; Claro, I.; et al. Colorectal cancers show distinct mutation spectra in members of the canonical WNT signaling pathway according to their anatomical location and type of genetic instability. Genes Chromosomes Cancer 2010, 49, 746–759. [Google Scholar]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta-catenin signalling thresholds for tumourigenesis. Oncogene 2012. [Google Scholar] [CrossRef]
- Watanabe, T.; Suda, T.; Tsunoda, T.; Uchida, N.; Ura, K.; Kato, T.; Hasegawa, S.; Satoh, S.; Ohgi, S.; Tahara, H.; et al. Identification of immunoglobulin superfamily 11 (IGSF11) as a novel target for cancer immunotherapy of gastrointestinal and hepatocellular carcinomas. Cancer Sci. 2005, 96, 498–506. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Munoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef]
- Joo, Y.E. Increased expression of brain-derived neurotrophic factor in irritable bowel syndrome and its correlation with abdominal pain (Gut 2012;61:685–694). J. Neurogastroenterol. Motil. 2013, 19, 109–111. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability, and autoimmunity: Celiac disease and type 1 diabetes paradigms. Ann. N. Y. Acad. Sci. 2009, 1165, 195–205. [Google Scholar] [CrossRef]
- Akil, H.; Perraud, A.; Melin, C.; Jauberteau, M.O.; Mathonnet, M. Fine-tuning roles of endogenous brain-derived neurotrophic factor, TrkB and sortilin in colorectal cancer cell survival. PLoS One 2011, 6, e25097. [Google Scholar]
- Enam, S.; Gan, D.D.; White, M.K.; del Valle, L.; Khalili, K. Regulation of human neurotropic JCV in colon cancer cells. Anticancer Res. 2006, 26, 833–841. [Google Scholar]
- Wang, X.; Tully, O.; Ngo, B.; Zitin, M.; Mullin, J.M. Epithelial tight junctional changes in colorectal cancer tissues. Sci. World J. 2011, 11, 826–841. [Google Scholar] [CrossRef]
- Soler, A.P.; Miller, R.D.; Laughlin, K.V.; Carp, N.Z.; Klurfeld, D.M.; Mullin, J.M. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 1999, 20, 1425–1431. [Google Scholar] [CrossRef]
- Grindedal, E.M.; Moller, P.; Eeles, R.; Stormorken, A.T.; Bowitz-Lothe, I.M.; Landro, S.M.; Clark, N.; Kvale, R.; Shanley, S.; Maehle, L. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2460–2467. [Google Scholar] [CrossRef]
- Desai, M.D.; Saroya, B.S.; Lockhart, A.C. Investigational therapies targeting the ErbB (EGFR, HER2, HER3, HER4) family in GI cancers. Expert Opin. Invest. Drugs 2013, 22, 341–356. [Google Scholar] [CrossRef]
- Khelwatty, S.A.; Essapen, S.; Seddon, A.M.; Modjtahedi, H. Prognostic significance and targeting of HER family in colorectal cancer. Front. Biosci. 2013, 18, 394–421. [Google Scholar] [CrossRef]
- Baraniskin, A.; Birkenkamp-Demtroder, K.; Maghnouj, A.; Zollner, H.; Munding, J.; Klein-Scory, S.; Reinacher-Schick, A.; Schwarte-Waldhoff, I.; Schmiegel, W.; Hahn, S.A. MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis 2012, 33, 732–739. [Google Scholar] [CrossRef]
- Bauer, K.M.; Hummon, A.B. Effects of the miR-143/-145 microRNA cluster on the colon cancer proteome and transcriptome. J. Proteome Res. 2012, 11, 4744–4754. [Google Scholar] [CrossRef]
- Cekaite, L.; Rantala, J.K.; Bruun, J.; Guriby, M.; Agesen, T.H.; Danielsen, S.A.; Lind, G.E.; Nesbakken, A.; Kallioniemi, O.; Lothe, R.A.; et al. MiR-9, -31, and -182 deregulation promote proliferation and tumor cell survival in colon cancer. Neoplasia 2012, 14, 868–879. [Google Scholar]
- Cheng, H.; Zhang, L.; Cogdell, D.E.; Zheng, H.; Schetter, A.J.; Nykter, M.; Harris, C.C.; Chen, K.; Hamilton, S.R.; Zhang, W. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One 2011, 6, e17745. [Google Scholar] [CrossRef]
- Dai, L.; Wang, W.; Zhang, S.; Jiang, Q.; Wang, R.; Dai, L.; Cheng, L.; Yang, Y.; Wei, Y.Q.; Deng, H.X. Vector-based miR-15a/16-1 plasmid inhibits colon cancer growth in vivo. Cell. Biol. Int. 2012, 36, 765–770. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- He, X.; Dong, Y.; Wu, C.W.; Zhao, Z.; Ng, S.S.; Chan, F.K.; Sung, J.J.; Yu, J. MicroRNA-218 inhibits cell cycle progression and promotes apoptosis in colon cancer by downregulating oncogene BMI-1. Mol. Med. 2012, 18, 1491–1498. [Google Scholar]
- Kenny, E.E.; Pe’er, I.; Karban, A.; Ozelius, L.; Mitchell, A.A.; Ng, S.M.; Erazo, M.; Ostrer, H.; Abraham, C.; Abreu, M.T.; et al. A genome-wide scan of Ashkenazi Jewish Crohn’s disease suggests novel susceptibility loci. PLoS Genet. 2012, 8, e1002559. [Google Scholar] [CrossRef]
- Migliore, C.; Martin, V.; Leoni, V.P.; Restivo, A.; Atzori, L.; Petrelli, A.; Isella, C.; Zorcolo, L.; Sarotto, I.; Casula, G.; et al. MiR-1 downregulation cooperates with MACC1 in promoting MET overexpression in human colon cancer. Clin. Cancer Res. 2012, 18, 737–747. [Google Scholar] [CrossRef]
- Nie, J.; Liu, L.; Zheng, W.; Chen, L.; Wu, X.; Xu, Y.; Du, X.; Han, W. MicroRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis 2012, 33, 220–225. [Google Scholar] [CrossRef]
- Okamoto, K.; Ishiguro, T.; Midorikawa, Y.; Ohata, H.; Izumiya, M.; Tsuchiya, N.; Sato, A.; Sakai, H.; Nakagama, H. MiR-493 induction during carcinogenesis blocks metastatic settlement of colon cancer cells in liver. EMBO J. 2012, 31, 1752–1763. [Google Scholar] [CrossRef]
- Okayama, H.; Schetter, A.J.; Harris, C.C. MicroRNAs and inflammation in the pathogenesis and progression of colon cancer. Dig. Dis. 2012, 30, 9–15. [Google Scholar] [CrossRef]
- Qased, A.B.; Yi, H.; Liang, N.; Ma, S.; Qiao, S.; Liu, X. MicroRNA-18a upregulates autophagy and ataxia telangiectasia mutated gene expression in HCT116 colon cancer cells. Mol. Med. Rep. 2013, 7, 559–564. [Google Scholar]
- Roy, S.; Levi, E.; Majumdar, A.P.; Sarkar, F.H. Expression of miR-34 is lost in colon cancer which can be re-expressed by a novel agent CDF. J. Hematol. Oncol. 2012, 5, 58. [Google Scholar] [CrossRef]
- Slaby, O.; Svoboda, M.; Michalek, J.; Vyzula, R. MicroRNAs in colorectal cancer: Translation of molecular biology into clinical application. Mol. Cancer 2009, 8, 102. [Google Scholar] [CrossRef]
- Strillacci, A.; Valerii, M.C.; Sansone, P.; Caggiano, C.; Sgromo, A.; Vittori, L.; Fiorentino, M.; Poggioli, G.; Rizzello, F.; Campieri, M.; et al. Loss of miR-101 expression promotes Wnt/beta-catenin signalling pathway activation and malignancy in colon cancer cells. J. Pathol. 2013, 229, 379–389. [Google Scholar] [CrossRef]
- Sun, J.Y.; Huang, Y.; Li, J.P.; Zhang, X.; Wang, L.; Meng, Y.L.; Yan, B.; Bian, Y.Q.; Zhao, J.; Wang, W.Z.; et al. MicroRNA-320a suppresses human colon cancer cell proliferation by directly targeting beta-catenin. Biochem. Biophys. Res. Commun. 2012, 420, 787–792. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Yang, Z.; Du, H.; Wu, Z.; Gong, J.; Yan, J.; Zheng, Q. MiR-145 regulates PAK4 via the MAPK pathway and exhibits an antitumor effect in human colon cells. Biochem. Biophys. Res. Commun. 2012, 427, 444–449. [Google Scholar] [CrossRef]
- Weissmann-Brenner, A.; Kushnir, M.; Lithwick Yanai, G.; Aharonov, R.; Gibori, H.; Purim, O.; Kundel, Y.; Morgenstern, S.; Halperin, M.; Niv, Y.; et al. Tumor microRNA-29a expression and the risk of recurrence in stage II colon cancer. Int. J. Oncol. 2012, 40, 2097–2103. [Google Scholar]
- Wu, J.; Ji, X.; Zhu, L.; Jiang, Q.; Wen, Z.; Xu, S.; Shao, W.; Cai, J.; Du, Q.; Zhu, Y.; et al. Up-regulation of microRNA-1290 impairs cytokinesis and affects the reprogramming of colon cancer cells. Cancer Lett. 2013, 329, 155–163. [Google Scholar] [CrossRef]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.F.; Zhang, X.J.; Xue, Y.F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2012, 33, 519–528. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, Z.; Lai, D.; Sun, J.; He, C.; Chu, Z.; Ye, H.; Chen, S.; Wang, J. MiR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br. J. Cancer 2012, 107, 352–359. [Google Scholar] [CrossRef]
- Zhu, R.; Yang, Y.; Tian, Y.; Bai, J.; Zhang, X.; Li, X.; Peng, Z.; He, Y.; Chen, L.; Pan, Q.; et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS One 2012, 7, e32170. [Google Scholar]
- Harada, O.; Suga, T.; Suzuki, T.; Nakamoto, K.; Kobayashi, M.; Nomiyama, T.; Nadano, D.; Ohyama, C.; Fukuda, M.N.; Nakayama, J. The role of trophinin, an adhesion molecule unique to human trophoblasts, in progression of colorectal cancer. Int. J. Cancer 2007, 121, 1072–1078. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Fukuda, Y.; Ohshima, K.; Terashima, M.; Yamaguchi, K.; Mochizuki, T. Placenta—Specific novel splice variants of Rho GDP dissociation inhibitor beta are highly expressed in cancerous cells. BMC Res. Notes 2012, 5, 666. [Google Scholar] [CrossRef]
- Wei, S.C.; Tsao, P.N.; Yu, S.C.; Shun, C.T.; Tsai-Wu, J.J.; Wu, C.H.; Su, Y.N.; Hsieh, F.J.; Wong, J.M. Placenta growth factor expression is correlated with survival of patients with colorectal cancer. Gut 2005, 54, 666–672. [Google Scholar] [CrossRef]
- Hrasovec, S.; Glavac, D. MicroRNAs as Novel Biomarkers in Colorectal Cancer. Front. Genet. 2012, 3, 180. [Google Scholar]
- Girirajan, S.; Eichler, E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010, 19, R176–R187. [Google Scholar] [CrossRef]
- Talseth-Palmer, B.A.; Holliday, E.G.; Evans, T.J.; McEvoy, M.; Attia, J.; Grice, D.M.; Masson, A.L.; Meldrum, C.; Spigelman, A.; Scott, R.J. Continuing difficulties in interpreting CNV data: lessons from a genome-wide CNV association study of Australian HNPCC/lynch syndrome patients. BMC Med. Genomics 2013, 6, 10. [Google Scholar] [CrossRef]
- Dellinger, A.E.; Saw, S.M.; Goh, L.K.; Seielstad, M.; Young, T.L.; Li, Y.J. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38, e105. [Google Scholar] [CrossRef]
- Tsuang, D.W.; Millard, S.P.; Ely, B.; Chi, P.; Wang, K.; Raskind, W.H.; Kim, S.; Brkanac, Z.; Yu, C.E. The effect of algorithms on copy number variant detection. PLoS One 2010, 5, e14456. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, Y.; Akula, N.; Alliey-Rodriguez, N.; Tang, J.; Bipolar Genome, S.; Gershon, E.S.; Liu, C. Accuracy of CNV detection from GWAS data. PLoS One 2011, 6, e14511. [Google Scholar] [CrossRef]
- Pinto, D.; Darvishi, K.; Shi, X.; Rajan, D.; Rigler, D.; Fitzgerald, T.; Lionel, A.C.; Thiruvahindrapuram, B.; Macdonald, J.R.; Mills, R.; et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011, 29, 512–520. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, J.H.; Chung, Y.J. Effect of combining multiple CNV defining algorithms on the reliability of CNV calls from snp genotyping data. Genomics Inform. 2012, 10, 194–199. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Masson, A.L.; Talseth-Palmer, B.A.; Evans, T.-J.; Grice, D.M.; Duesing, K.; Hannan, G.N.; Scott, R.J. Copy Number Variation in Hereditary Non-Polyposis Colorectal Cancer. Genes 2013, 4, 536-555. https://doi.org/10.3390/genes4040536
Masson AL, Talseth-Palmer BA, Evans T-J, Grice DM, Duesing K, Hannan GN, Scott RJ. Copy Number Variation in Hereditary Non-Polyposis Colorectal Cancer. Genes. 2013; 4(4):536-555. https://doi.org/10.3390/genes4040536
Chicago/Turabian StyleMasson, Amy L., Bente A. Talseth-Palmer, Tiffany-Jane Evans, Desma M. Grice, Konsta Duesing, Garry N. Hannan, and Rodney J. Scott. 2013. "Copy Number Variation in Hereditary Non-Polyposis Colorectal Cancer" Genes 4, no. 4: 536-555. https://doi.org/10.3390/genes4040536
APA StyleMasson, A. L., Talseth-Palmer, B. A., Evans, T.-J., Grice, D. M., Duesing, K., Hannan, G. N., & Scott, R. J. (2013). Copy Number Variation in Hereditary Non-Polyposis Colorectal Cancer. Genes, 4(4), 536-555. https://doi.org/10.3390/genes4040536