
Mapping Inherited Genetic Variation with Opposite Effects on Autoimmune Disease and Four Cancer Types Identifies Candidate Drug Targets Associated with the Anti-Tumor Immune Response

Abstract
1. Introduction
2. Materials and Methods
2.1. GWAS Data Sets
2.2. Meta-Analysis for Each Autoimmune/Autoinflammatory Disease and Cancer Pair
2.3. Identification of Immune-Related Genes Among Genes Nearest to Lead SNPs
2.4. Tumor Bulk and Single-Cell RNA-Seq Analyses to Prioritize Immune-Related Nearest Genes Based on Association with Intra-Tumor Immune Cell Infiltration
2.5. Functional Annotation to Link Prioritized Genes and Their Corresponding Lead SNPs
2.6. Evaluating the Druggability of Proteins Encoded by the Prioritized Genes
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | single nucleotide polymorphism |
| TCGA | The Cancer Genome Atlas |
| GWAS | genome-wide association study |
| PROTACs | proteolysis targeting chimeras |
| T1D | type 1 diabetes |
| RA | rheumatoid arthritis |
| HT | Hashimoto’s thyroiditis |
| MS | multiple sclerosis |
| SLE | systemic lupus erythematosus |
| UC | ulcerative colitis |
| CD | Crohn’s disease |
| ER | estrogen receptor |
| FUMA | Functional Mapping and Annotation |
| LD | linkage disequilibrium |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MSigDB | Molecular Signatures Database |
| TISCH2 | Tumor Immune Single-cell Hub 2 |
| eQTL | expression quantitative trait locus |
| RNA-Seq | ribonucleic acid-sequencing |
| RSEM | RNA-Seq by expectation maximization |
| OR | Odds ratio |
| CI | Confidence interval |
References
- Khan, Z.; Di Nucci, F.; Kwan, A.; Hammer, C.; Mariathasan, S.; Rouilly, V.; Carroll, J.; Fontes, M.; Ley Acosta, S.; Guardino, E.; et al. Polygenic Risk for Skin Autoimmunity Impacts Immune Checkpoint Blockade in Bladder Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 12288–12294. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Hammer, C.; Carroll, J.; Di Nucci, F.; Acosta, S.L.; Maiya, V.; Bhangale, T.; Hunkapiller, J.; Mellman, I.; Albert, M.L.; et al. Genetic Variation Associated with Thyroid Autoimmunity Shapes the Systemic Immune Response to PD-1 Checkpoint Blockade. Nat. Commun. 2021, 12, 3355. [Google Scholar] [CrossRef] [PubMed]
- Shahamatdar, S.; He, M.X.; Reyna, M.A.; Gusev, A.; AlDubayan, S.H.; Van Allen, E.M.; Ramachandran, S. Germline Features Associated with Immune Infiltration in Solid Tumors. Cell Rep. 2020, 30, 2900–2908.e4. [Google Scholar] [CrossRef]
- Sayaman, R.W.; Saad, M.; Thorsson, V.; Hu, D.; Hendrickx, W.; Roelands, J.; Porta-Pardo, E.; Mokrab, Y.; Farshidfar, F.; Kirchhoff, T.; et al. Germline Genetic Contribution to the Immune Landscape of Cancer. Immunity 2021, 54, 367–386.e8. [Google Scholar] [CrossRef]
- Liyanage, U.E.; MacGregor, S.; Bishop, D.T.; Shi, J.; An, J.; Ong, J.S.; Han, X.; Scolyer, R.A.; Martin, N.G.; Medland, S.E.; et al. Multi-Trait Genetic Analysis Identifies Autoimmune Loci Associated with Cutaneous Melanoma. J. Investig. Dermatol. 2022, 142, 1607–1616. [Google Scholar] [CrossRef]
- Fenaux, J.; Fang, X.; Huang, Y.-M.; Melero, C.; Bonnans, C.; Lowe, E.L.; Palumbo, T.; Lay, C.; Yi, Z.; Zhou, A.; et al. 23ME-00610, a Genetically Informed, First-in-Class Antibody Targeting CD200R1 to Enhance Antitumor T Cell Function. Oncoimmunology 2023, 12, 2217737. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and Hot Tumors: From Molecular Mechanisms to Targeted Therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association Analysis Identifies 65 New Breast Cancer Risk Loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Phelan, C.M.; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 New Susceptibility Loci for Different Histotypes of Epithelial Ovarian Cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association Analyses of More than 140,000 Men Identify 63 New Prostate Cancer Susceptibility Loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Bolla, M.K.; et al. Identification of Nine New Susceptibility Loci for Endometrial Cancer. Nat. Commun. 2018, 9, 3166. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.-G.; et al. Genome-Wide Association Study Implicates Immune Activation of Multiple Integrin Genes in Inflammatory Bowel Disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic Association Analyses Implicate Aberrant Regulation of Innate and Adaptive Immunity Genes in the Pathogenesis of Systemic Lupus Erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef]
- Saevarsdottir, S.; Olafsdottir, T.A.; Ivarsdottir, E.V.; Halldorsson, G.H.; Gunnarsdottir, K.; Sigurdsson, A.; Johannesson, A.; Sigurdsson, J.K.; Juliusdottir, T.; Lund, S.H.; et al. FLT3 Stop Mutation Increases FLT3 Ligand Level and Risk of Autoimmune Thyroid Disease. Nature 2020, 584, 619–623. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC); Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of Immune-Related Loci Identifies 48 New Susceptibility Variants for Multiple Sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar]
- Robertson, C.C.; Inshaw, J.R.J.; Onengut-Gumuscu, S.; Chen, W.-M.; Santa Cruz, D.F.; Yang, H.; Cutler, A.J.; Crouch, D.J.M.; Farber, E.; Bridges, S.L.; et al. Fine-Mapping, Trans-Ancestral and Genomic Analyses Identify Causal Variants, Cells, Genes and Drug Targets for Type 1 Diabetes. Nat. Genet. 2021, 53, 962–971. [Google Scholar] [CrossRef]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and Efficient Meta-Analysis of Genomewide Association Scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional Mapping and Annotation of Genetic Associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Stacey, D.; Fauman, E.B.; Ziemek, D.; Sun, B.B.; Harshfield, E.L.; Wood, A.M.; Butterworth, A.S.; Suhre, K.; Paul, D.S. ProGeM: A Framework for the Prioritization of Candidate Causal Genes at Molecular Quantitative Trait Loci. Nucleic Acids Res. 2019, 47, e3. [Google Scholar] [CrossRef] [PubMed]
- Chaussabel, D.; Baldwin, N. Democratizing Systems Immunology with Modular Transcriptional Repertoire Analyses. Nat. Rev. Immunol. 2014, 14, 271–280. [Google Scholar] [CrossRef]
- Li, S.; Rouphael, N.; Duraisingham, S.; Romero-Steiner, S.; Presnell, S.; Davis, C.; Schmidt, D.S.; Johnson, S.E.; Milton, A.; Rajam, G.; et al. Molecular Signatures of Antibody Responses Derived from a Systems Biology Study of Five Human Vaccines. Nat. Immunol. 2014, 15, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Y.; Dong, X.; Sun, D.; Liu, Z.; Yue, J.; Wang, H.; Li, T.; Wang, C. TISCH2: Expanded Datasets and New Tools for Single-Cell Transcriptome Analyses of the Tumor Microenvironment. Nucleic Acids Res. 2023, 51, D1425–D1431. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Mountjoy, E.; Carmona, M.; Peat, G.; Schmidt, E.M.; Hercules, A.; Fumis, L.; Miranda, A.; Carvalho-Silva, D.; Buniello, A.; et al. Open Targets Genetics: Systematic Identification of Trait-Associated Genes Using Large-Scale Genetics and Functional Genomics. Nucleic Acids Res. 2021, 49, D1311–D1320. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-eQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, N.; Spragins, E.; Kagda, M.S.; Li, M.; Assis, P.; Jolanki, O.; Luo, Y.; Cherry, J.M.; Boyle, A.P.; et al. Annotating and Prioritizing Human Non-Coding Variants with RegulomeDB v.2. Nat. Genet. 2023, 55, 724–726. [Google Scholar] [CrossRef]
- Raies, A.; Tulodziecka, E.; Stainer, J.; Middleton, L.; Dhindsa, R.S.; Hill, P.; Engkvist, O.; Harper, A.R.; Petrovski, S.; Vitsios, D. DrugnomeAI Is an Ensemble Machine-Learning Framework for Predicting Druggability of Candidate Drug Targets. Commun. Biol. 2022, 5, 1291. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hou, W.; Scharping, N.E.; Vendetti, F.P.; Srivastava, R.; Roy, C.N.; Menk, A.V.; Wang, Y.; Chauvin, J.-M.; Karukonda, P.; et al. IRF1 Inhibits Antitumor Immunity through the Upregulation of PD-L1 in the Tumor Cell. Cancer Immunol. Res. 2019, 7, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zheng, L.; Du, Q.; Yan, B.; Geller, D.A. Interferon Regulatory Factor 1 (IRF-1) and IRF-2 Regulate PD-L1 Expression in Hepatocellular Carcinoma (HCC) Cells. Cancer Immunol. Immunother. 2020, 69, 1891–1903. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, L.; Zou, Y.; Zhang, Y.; Zhang, M.; Xu, L.; Zheng, L.; He, W.; Yu, K.; Li, T.; et al. Disrupting the Phase Separation of KAT8-IRF1 Diminishes PD-L1 Expression and Promotes Antitumor Immunity. Nat. Cancer 2023, 4, 382–400. [Google Scholar] [CrossRef]
- Payne, K.J.; Dovat, S. Ikaros and Tumor Suppression in Acute Lymphoblastic Leukemia. Crit. Rev. Oncog. 2011, 16, 3–12. [Google Scholar] [CrossRef]
- Chen, J.C.; Perez-Lorenzo, R.; Saenger, Y.M.; Drake, C.G.; Christiano, A.M. IKZF1 Enhances Immune Infiltrate Recruitment in Solid Tumors and Susceptibility to Immunotherapy. Cell Syst. 2018, 7, 92–103.e4. [Google Scholar] [CrossRef]
- Feng, H.; Wang, T.; Ye, J.; Yang, Y.; Huang, X.; Lai, D.; Lv, Z.; Huang, Y.; Zhang, X. SPI1 Is a Prognostic Biomarker of Immune Infiltration and Immunotherapy Efficacy in Clear Cell Renal Cell Carcinoma. Discov. Oncol. 2022, 13, 134. [Google Scholar] [CrossRef]
- Luo, Q.; Dong, Z.; Xie, W.; Fu, X.; Lin, L.; Zeng, Q.; Chen, Y.; Ye, G.; Chen, M.; Hu, H.; et al. Apatinib Remodels the Immunosuppressive Tumor Ecosystem of Gastric Cancer Enhancing Anti-PD-1 Immunotherapy. Cell Rep. 2023, 42, 112437. [Google Scholar] [CrossRef]
- Willman, C.L. SH2B3: A New Leukemia Predisposition Gene. Blood 2013, 122, 2293–2295. [Google Scholar] [CrossRef]
- Wang, L.-N.; Zhang, Z.-T.; Wang, L.; Wei, H.-X.; Zhang, T.; Zhang, L.-M.; Lin, H.; Zhang, H.; Wang, S.-Q. TGF-Β1/SH2B3 Axis Regulates Anoikis Resistance and EMT of Lung Cancer Cells by Modulating JAK2/STAT3 and SHP2/Grb2 Signaling Pathways. Cell Death Dis. 2022, 13, 472. [Google Scholar] [CrossRef]
- Katayama, H.; Mori, T.; Seki, Y.; Anraku, M.; Iseki, M.; Ikutani, M.; Iwasaki, Y.; Yoshida, N.; Takatsu, K.; Takaki, S. Lnk Prevents Inflammatory CD8+ T-Cell Proliferation and Contributes to Intestinal Homeostasis. Eur. J. Immunol. 2014, 44, 1622–1632. [Google Scholar] [CrossRef]
- Pant, T.; Foda, B.; Geurts, A.; Chen, Y.-G. Lnk/Sh2b3 Modulates Bioenergetic Metabolism of Activated CD8 T Cells and Control the Development of Type 1 Diabetes. J. Immunol. 2023, 210, 77.03. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Joaquim, M.; Kuchel, G.A.; Ferrucci, L.; Harries, L.W.; Pilling, L.C.; Melzer, D. The Longevity-Associated SH2B3 (LNK) Genetic Variant: Selected Aging Phenotypes in 379,758 Subjects. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Turker, I.; Sharma, A.; Huang, S.; Johnson, D.B.; Alexander, M.R. Combination Immune Checkpoint Inhibitor Therapy Is Associated With Increased Blood Pressure in Melanoma Patients. Hypertension 2023, 80, e43–e45. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-L.; Shah, N.H.; Rubin, S.A.; Zhang, W.; Horkova, V.; Fallahee, I.R.; Stepanek, O.; Zon, L.I.; Kuriyan, J.; Weiss, A. Slow Phosphorylation of a Tyrosine Residue in LAT Optimizes T Cell Ligand Discrimination. Nat. Immunol. 2019, 20, 1481–1493. [Google Scholar] [CrossRef]
- Balagopalan, L.; Ashwell, B.A.; Bernot, K.M.; Akpan, I.O.; Quasba, N.; Barr, V.A.; Samelson, L.E. Enhanced T-Cell Signaling in Cells Bearing Linker for Activation of T-Cell (LAT) Molecules Resistant to Ubiquitylation. Proc. Natl. Acad. Sci. USA 2011, 108, 2885–2890. [Google Scholar] [CrossRef]
- Kent, A.; Longino, N.V.; Christians, A.; Davila, E. Naturally Occurring Genetic Alterations in Proximal TCR Signaling and Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 658611. [Google Scholar] [CrossRef]
- Charlton, B.; Lafferty, K.J. The Th1/Th2 Balance in Autoimmunity. Curr. Opin. Immunol. 1995, 7, 793–798. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L. Tumor Antigen-Specific T Helper Cells in Cancer Immunity and Immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef]
- Ellyard, J.I.; Simson, L.; Parish, C.R. Th2-Mediated Anti-Tumour Immunity: Friend or Foe? Tissue Antigens 2007, 70, 1–11. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the Immune Response to Cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Hollande, C.; Boussier, J.; Ziai, J.; Nozawa, T.; Bondet, V.; Phung, W.; Lu, B.; Duffy, D.; Paradis, V.; Mallet, V.; et al. Inhibition of the Dipeptidyl Peptidase DPP4 (CD26) Reveals IL-33-Dependent Eosinophil-Mediated Control of Tumor Growth. Nat. Immunol. 2019, 20, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Fatumo, S.; Chikowore, T.; Choudhury, A.; Ayub, M.; Martin, A.R.; Kuchenbaecker, K. A Roadmap to Increase Diversity in Genomic Studies. Nat. Med. 2022, 28, 243–250. [Google Scholar] [CrossRef]
- Groha, S.; Alaiwi, S.A.; Xu, W.; Naranbhai, V.; Nassar, A.H.; Bakouny, Z.; El Zarif, T.; Saliby, R.M.; Wan, G.; Rajeh, A.; et al. Germline Variants Associated with Toxicity to Immune Checkpoint Blockade. Nat. Med. 2022, 28, 2584–2591. [Google Scholar] [CrossRef]
- Kinnersley, B.; Sud, A.; Coker, E.A.; Tym, J.E.; Di Micco, P.; Al-Lazikani, B.; Houlston, R.S. Leveraging Human Genetics to Guide Cancer Drug Development. JCO Clin. Cancer Inform. 2018, 2, 1–11. [Google Scholar] [CrossRef]
- Minikel, E.V.; Painter, J.L.; Dong, C.C.; Nelson, M.R. Refining the Impact of Genetic Evidence on Clinical Success. Nature 2024, 629, 624–629. [Google Scholar] [CrossRef]
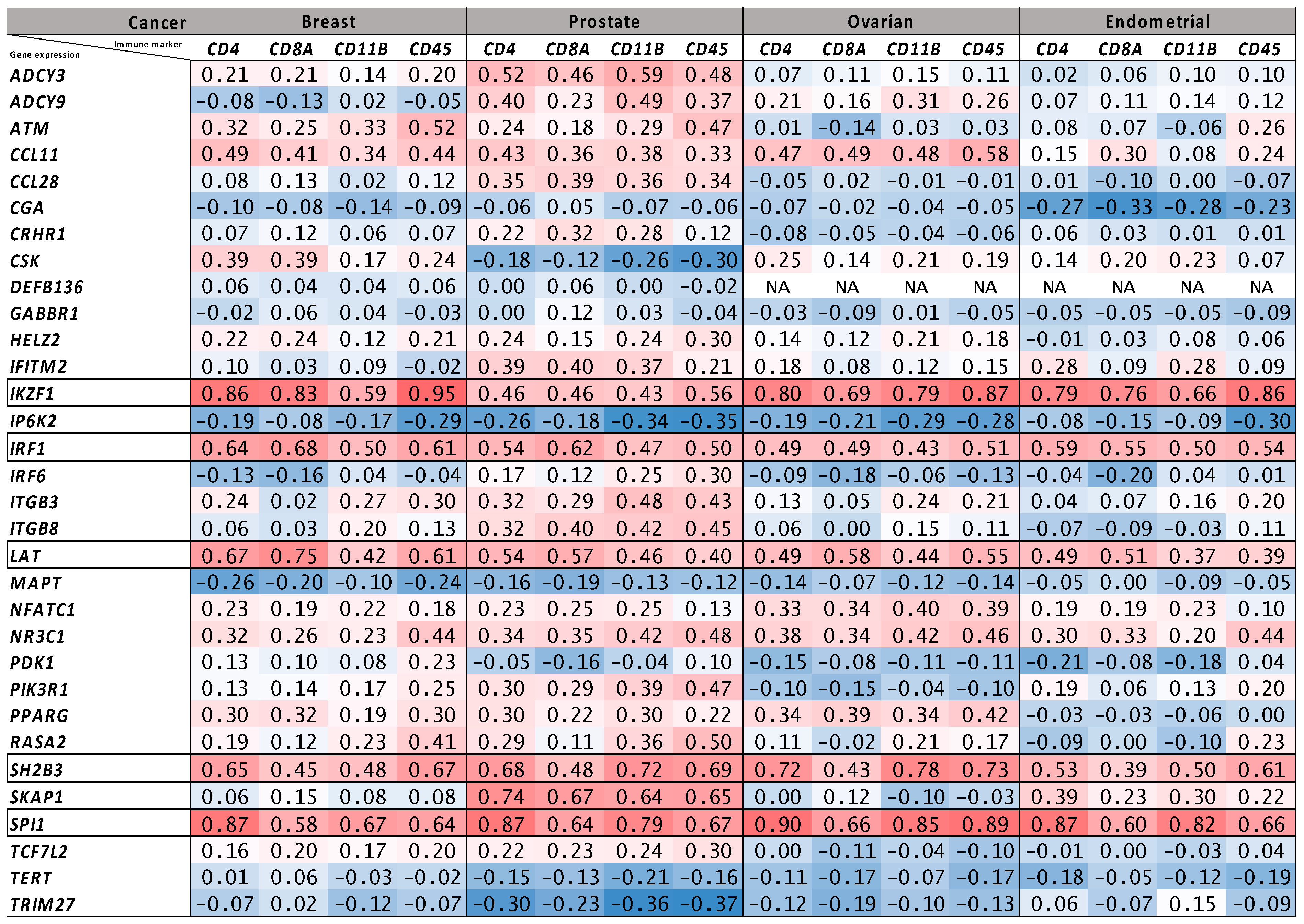
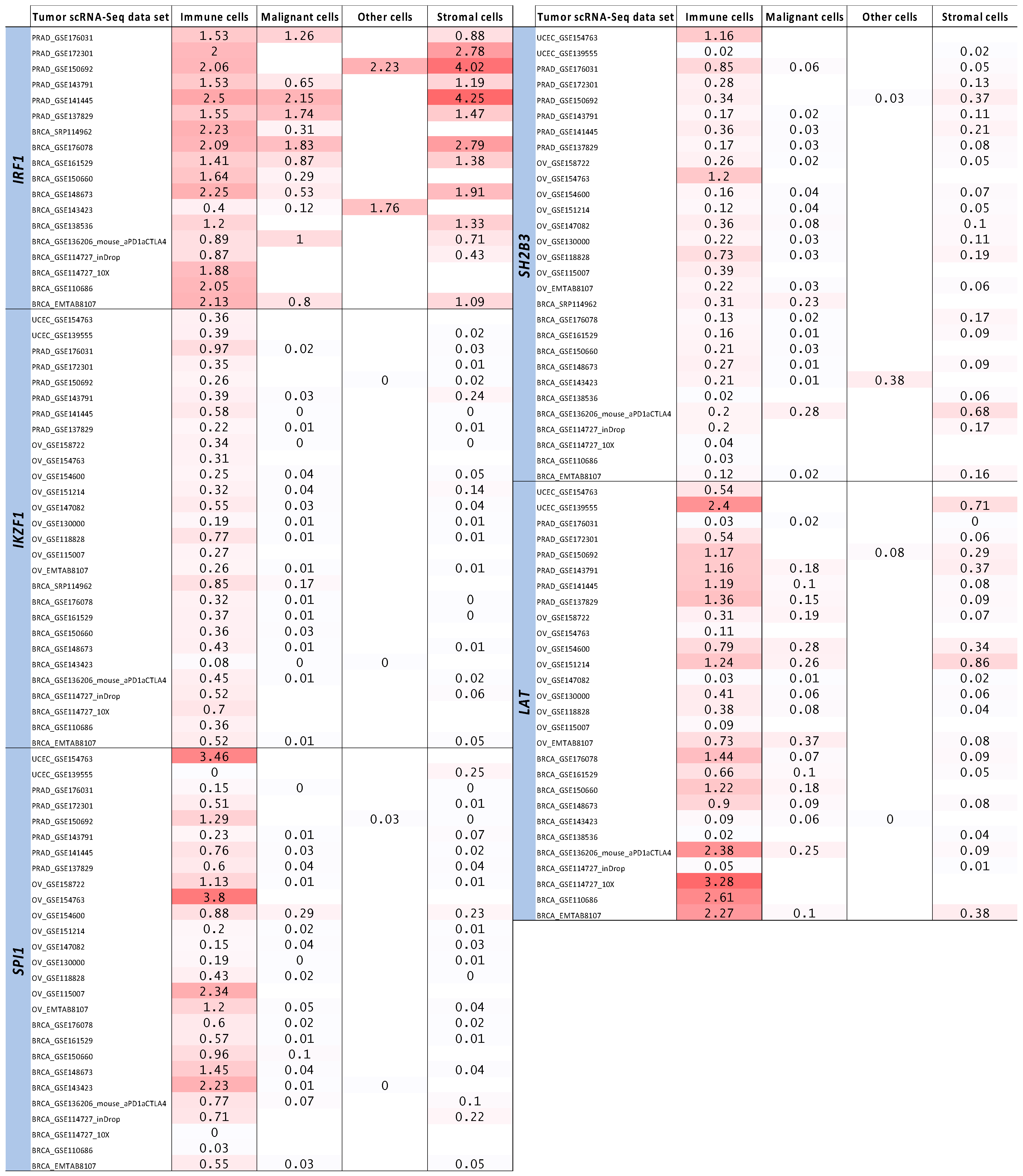


{kind=link}
{kind=link}
{kind=link}
| Genes | ImmPort | Enrichr | ImmPort Category/Enrichr Pathway |
|---|---|---|---|
| ADCY3 | ✓ | Human T-cell leukemia virus 1 infection | |
| ADCY9 | ✓ | Human T-cell leukemia virus 1 infection | |
| ATM | ✓ | Human T-cell leukemia virus 1 infection | |
| CCL11 | ✓ | Cytokines/Antimicrobials/Chemokines | |
| CCL28 | ✓ | Cytokines/Antimicrobials/Chemokines | |
| CGA | ✓ | Cytokines | |
| CRHR1 | ✓ | Cytokine receptors | |
| CSK | ✓ | Antimicrobials | |
| DEFB136 | ✓ | Antimicrobials | |
| GABBR1 | ✓ | Inflammatory Response | |
| HELZ2 | ✓ | Interferon α Response | |
| IFITM2 | ✓ | Interferon α/β signaling R-HSA-909733/Interferon α Response | |
| IKZF1 | ✓ | Development of pulmonary dendritic cells and macrophage subsets WP3892 | |
| IP6K2 | ✓ | Interferon α/β signaling R-HSA-909733 | |
| IRF1 | ✓ ** | ✓ ** | Antimicrobials/Interferon α/β signaling R-HSA-909733/Interferon α Response |
| IRF6 | ✓ | Interferon α/β signaling R-HSA-909733/Inflammatory Response | |
| ITGB3 | ✓ | Inflammatory Response | |
| ITGB8 | ✓ | Inflammatory Response | |
| LAT | ✓ ** | ✓ ** | Natural killer cell cytotoxicity/TCR signaling pathway/Modulators of TCR signaling and T cell activation WP5072 |
| MAPT | ✓ | Antimicrobials | |
| NFATC1 | ✓ ** | ✓ ** | Natural killer cell cytotoxicity/TCR signaling pathway/BCR signaling pathway/Human T-cell leukemia virus 1 infection |
| NR3C1 | ✓ | Cytokine receptors | |
| PDK1 | ✓ | TCR signaling pathway | |
| PIK3R1 | ✓ ** | ✓ ** | Natural killer cell cytotoxicity/TCR signaling pathway/BCR signaling pathway/Modulators of TCR signaling and T cell activation WP5072/Human T-cell leukemia virus 1 infection |
| PPARG | ✓ | Cytokine receptors/Antimicrobials | |
| RASA2 | ✓ | Natural killer cell cytotoxicity/TCR signaling pathway/BCR signaling pathway/Modulators of TCR signaling and T cell activation WP5072/Human T-cell leukemia virus 1 infection | |
| SH2B3 | ✓ | TCR signaling pathway/Modulators of TCR signaling and T cell activation WP5072 | |
| SKAP1 | ✓ | TCR signaling pathway | |
| SPI1 | ✓ | Development of pulmonary dendritic cells and macrophage subsets WP3892/Human T-cell leukemia virus 1 infection | |
| TCF7L2 | ✓ | Antimicrobials | |
| TERT | ✓ | Human T-cell leukemia virus 1 infection | |
| TRIM27 | ✓ | Antimicrobials |
| OpenTargets Lead SNP-to-Gene Evidence | DrugnomeAI Percentile Scores for Druggability | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lead SNP | Gene | sQTL | eQTL | PCHi-C | VEP | Oncology Specific-Antibody | Oncology Specific-Small Molecule | Small Molecule | Antibody | PROTAC |
| rs2070721 | IRF1 | Yes | Yes | intronic variant | 92 | 76 | 75 | 95 | 91 | |
| rs10230978 | IKZF1 | Yes | Yes | 89 | 87 | 85 | 88 | 93 | ||
| rs3740688 | SPI1 | Yes | missense variant | 96 | 82 | 81 | 93 | 88 | ||
| rs3184504 | SH2B3 | Yes | Yes | 94 | 89 | 89 | 96 | 95 | ||
| rs4788115 | LAT | Yes | intronic variant | 97 | 85 | 84 | 96 | 95 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Epstein, M.P.; Schildkraut, J.M.; Kar, S.P. Mapping Inherited Genetic Variation with Opposite Effects on Autoimmune Disease and Four Cancer Types Identifies Candidate Drug Targets Associated with the Anti-Tumor Immune Response. Genes 2025, 16, 575. https://doi.org/10.3390/genes16050575
Chen J, Epstein MP, Schildkraut JM, Kar SP. Mapping Inherited Genetic Variation with Opposite Effects on Autoimmune Disease and Four Cancer Types Identifies Candidate Drug Targets Associated with the Anti-Tumor Immune Response. Genes. 2025; 16(5):575. https://doi.org/10.3390/genes16050575
Chicago/Turabian StyleChen, Junyu, Michael P. Epstein, Joellen M. Schildkraut, and Siddhartha P. Kar. 2025. "Mapping Inherited Genetic Variation with Opposite Effects on Autoimmune Disease and Four Cancer Types Identifies Candidate Drug Targets Associated with the Anti-Tumor Immune Response" Genes 16, no. 5: 575. https://doi.org/10.3390/genes16050575
APA StyleChen, J., Epstein, M. P., Schildkraut, J. M., & Kar, S. P. (2025). Mapping Inherited Genetic Variation with Opposite Effects on Autoimmune Disease and Four Cancer Types Identifies Candidate Drug Targets Associated with the Anti-Tumor Immune Response. Genes, 16(5), 575. https://doi.org/10.3390/genes16050575