
Dysregulation of Locus-Specific Repetitive Elements in TCGA Pan-Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Repetitive Elements (REs) and Their Regulations in the Human Genome
1.2. Dysregulation of REs in Cancer Genomes
2. Materials and Methods
2.1. Determine Differentially Expressed REs at Locus-Specific Levels Across 12 Cancer Types
2.2. Determine Differentially Methylated REs at Locus-Specific Levels Across 12 Cancer Types
2.3. Determine the Association Between RE Methylation and Expression Changes at the Locus-Specific Level
2.4. Statistical Analysis
3. Results
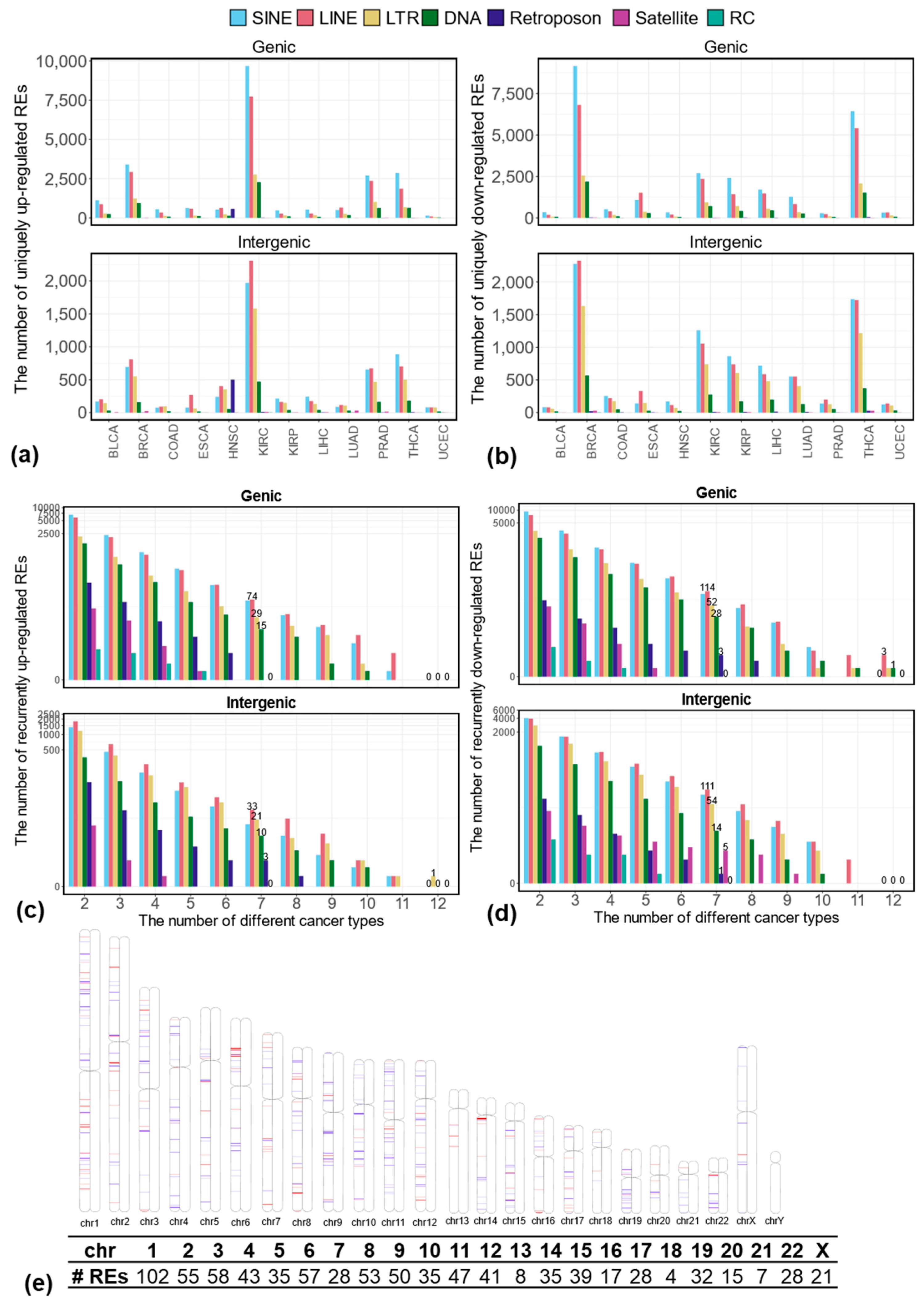
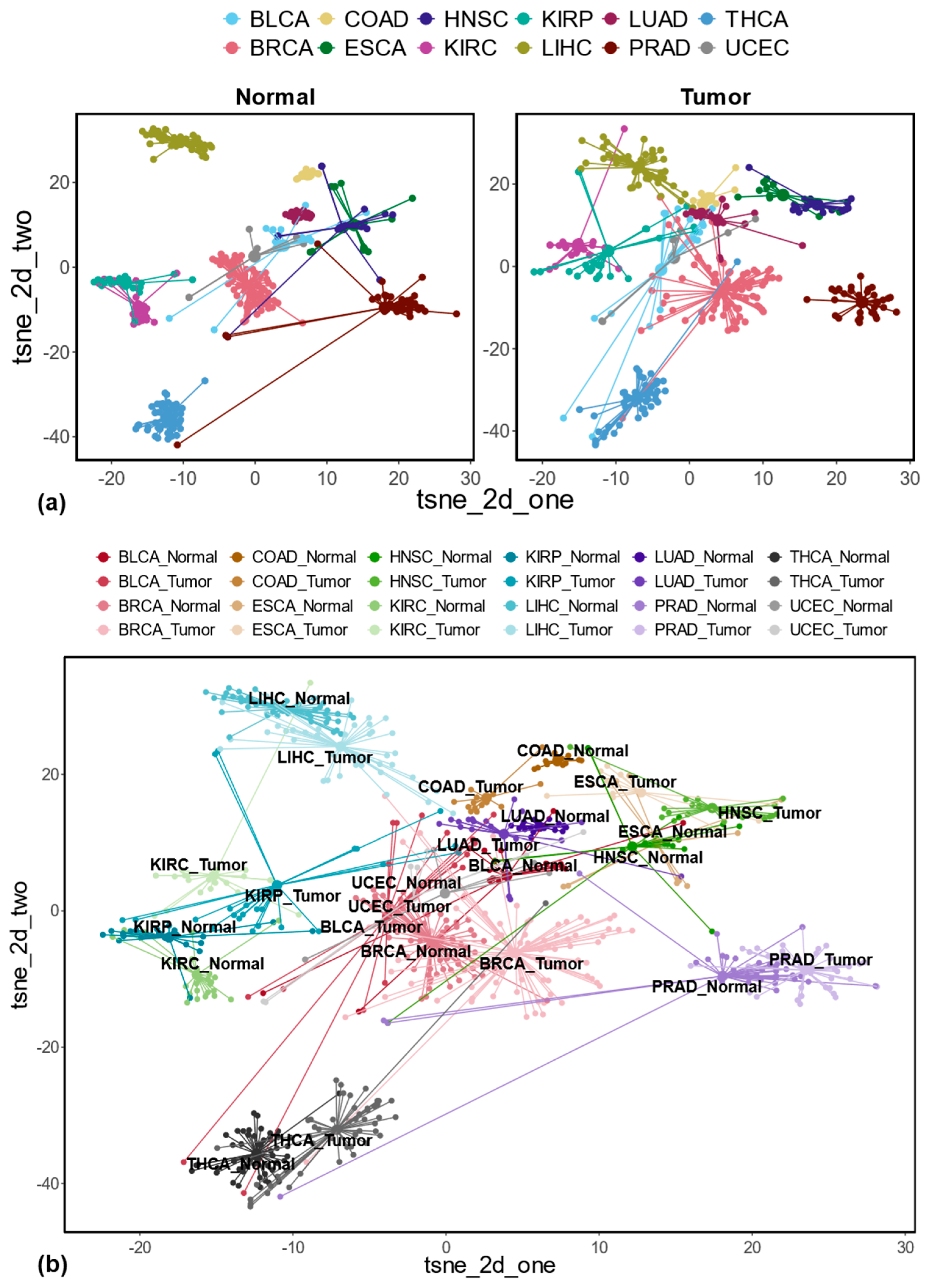
3.1. Differentially Expressed REs Between Tumor and Matched Normal Samples at the Locus-Specific Level
3.2. Uniquely Differentially Expressed REs in Each Cancer Type at the Locus-Specific Level
3.3. Recurrently Differentially Expressed REs in Multiple Cancer Types at the Locus-Specific Level
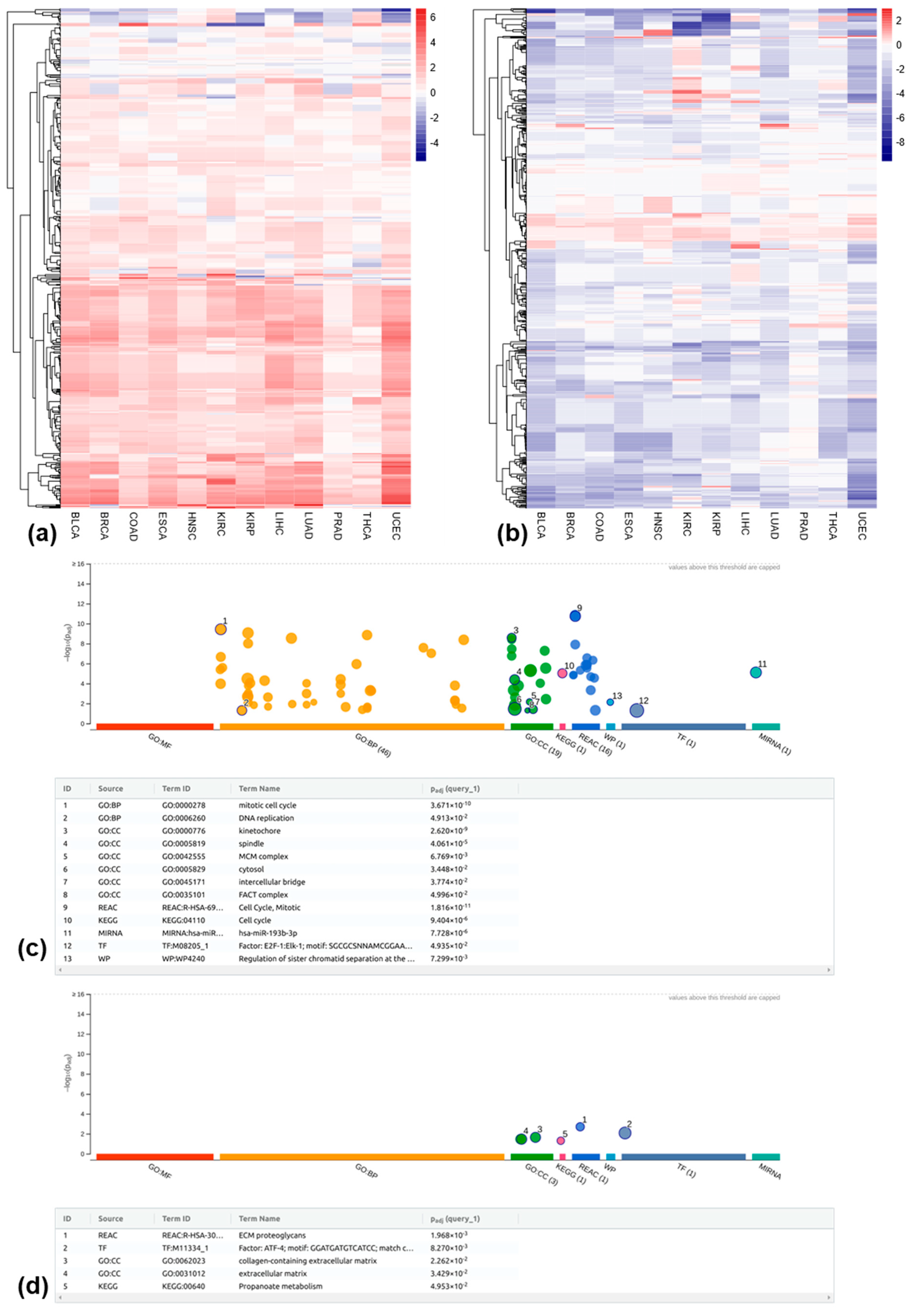
3.4. Differentially Methylated REs Between Tumor and Matched Normal Samples at the Locus-Specific Level
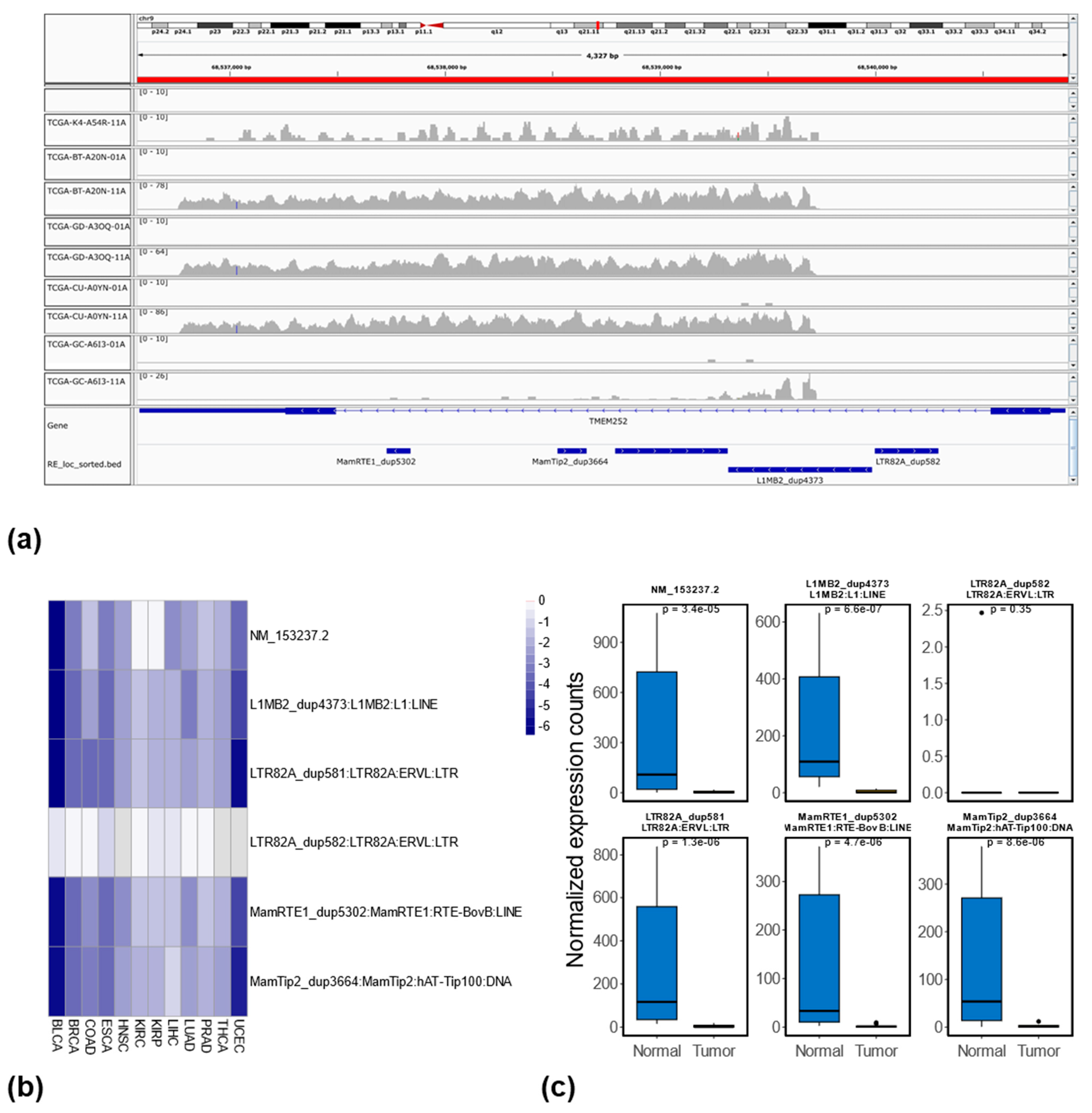
3.5. Relationship Between RE Methylation and Expression at the Locus-Specific Level
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodić, N.; Burns, K.H. Long Interspersed Element–1 (LINE-1): Passenger or Driver in Human Neoplasms? PLoS Genet. 2013, 9, e1003402. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013. Available online: https://www.repeatmasker.org/ (accessed on 20 November 2023).
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Thakur, J.; Packiaraj, J.; Henikoff, S. Sequence, Chromatin and Evolution of Satellite DNA. Int. J. Mol. Sci. 2021, 22, 4309. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA sequence detection and its role in the human genome. Commun. Biol. 2023, 6, 954. [Google Scholar] [CrossRef]
- Lerat, E.; Casacuberta, J.; Chaparro, C.; Vieira, C. On the Importance to Acknowledge Transposable Elements in Epigenomic Analyses. Genes 2019, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Barro-Trastoy, D.; Köhler, C. Helitrons: Genomic parasites that generate developmental novelties. Trends Genet. 2024, 40, 437–448. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Cajuso, T.; Sulo, P.; Tanskanen, T.; Katainen, R.; Taira, A.; Hänninen, U.A.; Kondelin, J.; Forsström, L.; Välimäki, N.; Aavikko, M.; et al. Retrotransposon insertions can initiate colorectal cancer and are associated with poor survival. Nat. Commun. 2019, 10, 4022. [Google Scholar] [CrossRef]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable Elements: An Abundant and Natural Source of Regulatory Sequences for Host Genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef]
- Levine, A.J.; Ting, D.T.; Greenbaum, B.D. P53 and the defenses against genome instability caused by transposons and repetitive elements. BioEssays 2016, 38, 508–513. [Google Scholar] [CrossRef]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin 2021, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2010; Volume 70, pp. 27–56. ISBN 978-0-12-380866-0. [Google Scholar]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef]
- Kanholm, T.; Rentia, U.; Hadley, M.; Karlow, J.A.; Cox, O.L.; Diab, N.; Bendall, M.L.; Dawson, T.; McDonald, J.I.; Xie, W.; et al. Oncogenic Transformation Drives DNA Methylation Loss and Transcriptional Activation at Transposable Element Loci. Cancer Res. 2023, 83, 2584–2599. [Google Scholar] [CrossRef]
- Choi, S.H.; Worswick, S.; Byun, H.; Shear, T.; Soussa, J.C.; Wolff, E.M.; Douer, D.; Garcia-Manero, G.; Liang, G.; Yang, A.S. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int. J. Cancer 2009, 125, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liang, C. The insertion and dysregulation of transposable elements in osteosarcoma and their association with patient event-free survival. Sci. Rep. 2022, 12, 377. [Google Scholar] [CrossRef]
- Kolbe, A.R.; Bendall, M.L.; Pearson, A.T.; Paul, D.; Nixon, D.F.; Pérez-Losada, M.; Crandall, K.A. Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival. Viruses 2020, 12, 956. [Google Scholar] [CrossRef]
- Steiner, M.C.; Marston, J.L.; Iñiguez, L.P.; Bendall, M.L.; Chiappinelli, K.B.; Nixon, D.F.; Crandall, K.A. Locus-Specific Characterization of Human Endogenous Retrovirus Expression in Prostate, Breast, and Colon Cancers. Cancer Res. 2021, 81, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 7, 10–12. [Google Scholar] [CrossRef]
- Andrews, S.; FastQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 November 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Pedregosa, F. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Rishishwar, L.; Conley, A.B.; Wigington, C.H.; Wang, L.; Valderrama-Aguirre, A.; King Jordan, I. Ancestry, admixture and fitness in Colombian genomes. Sci. Rep. 2015, 5, 12376. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genomics 2017, 44, 119–121. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2015. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 25 November 2023).
- Negri, G.L.; Grande, B.M.; Delaidelli, A.; El-Naggar, A.; Cochrane, D.; Lau, C.C.; Triche, T.J.; Moore, R.A.; Jones, S.J.; Montpetit, A.; et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J. Pathol. 2019, 249, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Futscher, B.W. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics 2018, 13, 61–72. [Google Scholar] [CrossRef]
- Fan, S.; Tang, J.; Li, N.; Zhao, Y.; Ai, R.; Zhang, K.; Wang, M.; Du, W.; Wang, W. Integrative analysis with expanded DNA methylation data reveals common key regulators and pathways in cancers. Npj Genom. Med. 2019, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K. IlluminaHumanMethylation450kanno.ilmn12.hg19: Annotation for Illumina’s 450k Methylation Arrays. 2016. Available online: https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylation450kanno.ilmn12.hg19.html (accessed on 5 February 2024).
- Ando, M.; Saito, Y.; Xu, G.; Bui, N.Q.; Medetgul-Ernar, K.; Pu, M.; Fisch, K.; Ren, S.; Sakai, A.; Fukusumi, T.; et al. Chromatin dysregulation and DNA methylation at transcription start sites associated with transcriptional repression in cancers. Nat. Commun. 2019, 10, 2188. [Google Scholar] [CrossRef]
- Dale, R.K.; Pedersen, B.S.; Quinlan, A.R. Pybedtools: A flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 2011, 27, 3423–3424. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Alboukadel Kassambara. Ggpubr: “ggplot2” Based Publication Ready Plots. 2020. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 5 February 2024).
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Bendall, M.L.; De Mulder, M.; Iñiguez, L.P.; Lecanda-Sánchez, A.; Pérez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Terán, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef]
- Savytska, N.; Heutink, P.; Bansal, V. Transcription start site signal profiling improves transposable element RNA expression analysis at locus-level. Front. Genet. 2022, 13, 1026847. [Google Scholar] [CrossRef] [PubMed]
- Reggiardo, R.E.; Maroli, S.V.; Peddu, V.; Davidson, A.E.; Hill, A.; LaMontagne, E.; Aaraj, Y.A.; Jain, M.; Chan, S.Y.; Kim, D.H. Profiling of repetitive RNA sequences in the blood plasma of patients with cancer. Nat. Biomed. Eng. 2023, 7, 1627–1635. [Google Scholar] [CrossRef]
- Talatam, A.; Reddy, P.K.; Motohashi, N.; Vanam, A.; Gollapudi, R. Targeting Overexpressed Cyclin Dependent Kinase 1 (CDK1) in Human Cancers: Kamalachalcone A Emerged as Potential Inhibitor of CDK1 Kinase Through in Silico Docking Study. Oncogen 2023, 6, 25. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, D.; Bai, Y.; Zhang, Y.; Zheng, Z.; Fu, Q.; Yi, B.; Jiang, Y.; Zhang, Z.; Zhu, J. ESCO2’s oncogenic role in human tumors: A pan-cancer analysis and experimental validation. BMC Cancer 2024, 24, 452. [Google Scholar] [CrossRef]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar] [CrossRef]
- Wu, X.; Wang, H.; Lian, Y.; Chen, L.; Gu, L.; Wang, J.; Huang, Y.; Deng, M.; Gao, Z.; Huang, Y. GTSE1 promotes cell migration and invasion by regulating EMT in hepatocellular carcinoma and is associated with poor prognosis. Sci. Rep. 2017, 7, 5129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xing, Z.; Xiao, Y.; Li, M.; Li, X.; Wang, D.; Dong, Z. The Value of H2BC12 for Predicting Poor Survival Outcomes in Patients With WHO Grade II and III Gliomas. Front. Mol. Biosci. 2022, 9, 816939. [Google Scholar] [CrossRef]
- Han, J.; Wang, F.; Lan, Y.; Wang, J.; Nie, C.; Liang, Y.; Song, R.; Zheng, T.; Pan, S.; Pei, T.; et al. KIFC1 regulated by miR-532-3p promotes epithelial-to-mesenchymal transition and metastasis of hepatocellular carcinoma via gankyrin/AKT signaling. Oncogene 2019, 38, 406–420. [Google Scholar] [CrossRef]
- Nguyen, M.-H.; Ueda, K.; Nakamura, Y.; Daigo, Y. Identification of a novel oncogene, MMS22L, involved in lung and esophageal carcinogenesis. Int. J. Oncol. 2012, 41, 1285–1296. [Google Scholar] [CrossRef]
- Mo, J.; Moye, S.L.; McKay, R.M.; Le, L.Q. Neurofibromin and suppression of tumorigenesis: Beyond the GAP. Oncogene 2022, 41, 1235–1251. [Google Scholar] [CrossRef]
- Lin, C.; Chung, M.; Chen, W.; Chien, C. Growth inhibitory effect of the human NIT2 gene and its allelic imbalance in cancers. FEBS J. 2007, 274, 2946–2956. [Google Scholar] [CrossRef]
- Onagoruwa, O.T.; Pal, G.; Ochu, C.; Ogunwobi, O.O. Oncogenic Role of PVT1 and Therapeutic Implications. Front. Oncol. 2020, 10, 17. [Google Scholar] [CrossRef]
- Wang, W.; Lopez McDonald, M.C.; Kim, C.; Ma, M.; Pan, Z. (Tommy); Kaufmann, C.; Frank, D.A. The complementary roles of STAT3 and STAT1 in cancer biology: Insights into tumor pathogenesis and therapeutic strategies. Front. Immunol. 2023, 14, 1265818. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, S.; Wang, Y.; Liu, J.; Wang, X. UBE2C promotes the proliferation of acute myeloid leukemia cells through PI3K/AKT activation. BMC Cancer 2024, 24, 497. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhou, B.; Zhang, J.; Geng, P.; Liu, K.; Zhu, Y.; Zhu, W. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumor Biol. 2014, 35, 7935–7944. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.-C.; Cheng, C.-F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, Y.; Yu, H.; Shen, B.; Liang, Y.; Jin, R.; Liu, X.; Shi, L.; Cai, X. Role of DUSP1/MKP1 in tumorigenesis, tumor progression and therapy. Cancer Med. 2016, 5, 2061–2068. [Google Scholar] [CrossRef]
- Hirata, D.; Yamabuki, T.; Miki, D.; Ito, T.; Tsuchiya, E.; Fujita, M.; Hosokawa, M.; Chayama, K.; Nakamura, Y.; Daigo, Y. Involvement of Epithelial Cell Transforming Sequence-2 Oncoantigen in Lung and Esophageal Cancer Progression. Clin. Cancer Res. 2009, 15, 256–266. [Google Scholar] [CrossRef]
- Ahmat Amin, M.K.B.; Shimizu, A.; Zankov, D.P.; Sato, A.; Kurita, S.; Ito, M.; Maeda, T.; Yoshida, T.; Sakaue, T.; Higashiyama, S.; et al. Epithelial membrane protein 1 promotes tumor metastasis by enhancing cell migration via copine-III and Rac1. Oncogene 2018, 37, 5416–5434. [Google Scholar] [CrossRef]
- Heinrich, R.; Livne, E.; Ben-Izhak, O.; Aronheim, A. The c-Jun Dimerization Protein 2 Inhibits Cell Transformation and Acts as a Tumor Suppressor Gene. J. Biol. Chem. 2004, 279, 5708–5715. [Google Scholar] [CrossRef] [PubMed]
- DiFeo, A.; Martignetti, J.A.; Narla, G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist. Updat. 2009, 12, 1–7. [Google Scholar] [CrossRef]
- Von Karstedt, S. NDRG2 programs tumor-associated macrophages for tumor support. Cell Death Dis. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Chen, B.; Huo, J.; Liu, X. Therapeutic potential of NR4A1 in cancer: Focus on metabolism. Front. Oncol. 2022, 12, 972984. [Google Scholar] [CrossRef]
- Choi, Y.M.; Kim, K.B.; Lee, J.H.; Chun, Y.K.; An, I.S.; An, S.; Bae, S. DBC2/RhoBTB2 functions as a tumor suppressor protein via Musashi-2 ubiquitination in breast cancer. Oncogene 2017, 36, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Z.; Li, W.; Zhang, S. SOCS2 is a potential prognostic marker that suppresses the viability of hepatocellular carcinoma cells. Oncol. Lett. 2021, 21, 399. [Google Scholar] [CrossRef]
- Li, T.; Liu, X.; Yang, A.; Fu, W.; Yin, F.; Zeng, X. Associations of tumor suppressor SPARCL1 with cancer progression and prognosis. Oncol. Lett. 2017, 14, 2603–2610. [Google Scholar] [CrossRef]
- Jaafar, L.; Chamseddine, Z.; El-Sibai, M. StarD13: A potential star target for tumor therapeutics. Hum. Cell 2020, 33, 437–443. [Google Scholar] [CrossRef]
- Zheng, Z.; Song, Y. Synaptopodin-2: A potential tumor suppressor. Cancer Cell Int. 2023, 23, 158. [Google Scholar] [CrossRef]
- Zhao, Z.; Lu, L.; Li, W. TAGLN2 promotes the proliferation, invasion, migration and epithelial-mesenchymal transition of colorectal cancer cells by activating STAT3 signaling through ANXA2. Oncol. Lett. 2021, 22, 737. [Google Scholar] [CrossRef]
- Su, C.-W.; Lin, C.-W.; Yang, W.-E.; Yang, S.-F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Wu, Z.; Zhang, X.; Zeng, T.; Dai, W.; Qiu, N.; Xu, M.; Qiao, Y.; Ke, L.; Zhao, J.; et al. TMEM25 inhibits monomeric EGFR-mediated STAT3 activation in basal state to suppress triple-negative breast cancer progression. Nat. Commun. 2023, 14, 2342. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, R.; Wang, L.; Fu, G.; Zhang, C.; Zhang, Y.; Yu, J. TMEM252 inhibits epithelial–mesenchymal transition and progression in papillary thyroid carcinoma by regulating Notch1 expression. Head Neck 2024, 47, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Le, T.N.; Miyazaki, Y.; Takuno, S.; Saze, H. Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 3911–3921. [Google Scholar] [CrossRef]
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef]
- Saze, H. Epigenetic regulation of intragenic transposable elements: A two-edged sword. J. Biochem. 2018, 164, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Rech, G.E.; Radío, S.; Guirao-Rico, S.; Aguilera, L.; Horvath, V.; Green, L.; Lindstadt, H.; Jamilloux, V.; Quesneville, H.; González, J. Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila. Nat. Commun. 2022, 13, 1948. [Google Scholar] [CrossRef]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648–5657. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Liang, C. Dysregulation of Locus-Specific Repetitive Elements in TCGA Pan-Cancers. Genes 2025, 16, 528. https://doi.org/10.3390/genes16050528
Wang C, Liang C. Dysregulation of Locus-Specific Repetitive Elements in TCGA Pan-Cancers. Genes. 2025; 16(5):528. https://doi.org/10.3390/genes16050528
Chicago/Turabian StyleWang, Chao, and Chun Liang. 2025. "Dysregulation of Locus-Specific Repetitive Elements in TCGA Pan-Cancers" Genes 16, no. 5: 528. https://doi.org/10.3390/genes16050528
APA StyleWang, C., & Liang, C. (2025). Dysregulation of Locus-Specific Repetitive Elements in TCGA Pan-Cancers. Genes, 16(5), 528. https://doi.org/10.3390/genes16050528