
Integrating Artificial Intelligence and Bioinformatics Methods to Identify Disruptive STAT1 Variants Impacting Protein Stability and Function

Abstract
1. Introduction
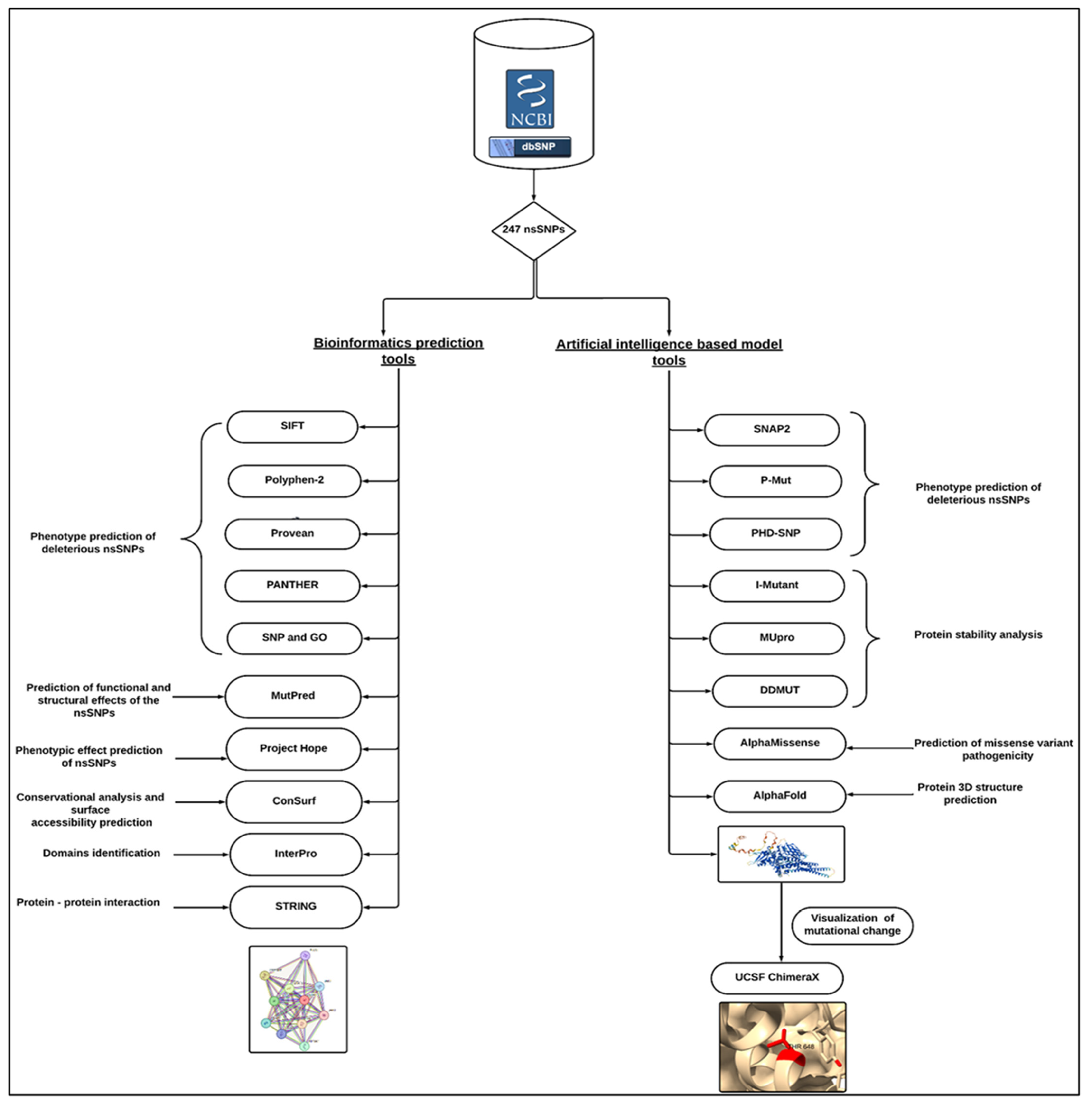
2. Materials and Method
2.1. Data Retrieval
2.2. Phenotype Prediction of Deleterious ns SNPs
2.3. Predicting Functional and Structural Effects of the nsSNP
2.4. Protein Stability Analysis of Predicted STAT1 nsSNPs
2.5. Prediction of Missense Variant Pathogenicity
2.6. Three-Dimensional Structure Prediction and Visualization
2.7. Phenotypic Effects Prediction
2.8. Conservational Analysis and Surface Accessibility Prediction of STAT1
2.9. Identification of nsSNPs in STAT1 Protein Domains
2.10. Prediction of Protein–Protein Interactions
3. Results
3.1. Distribution of STAT1 Gene SNP Datasets
3.2. Identification of Deleterious Missense Mutation
3.3. MutPred Prediction for Functional and Structural Modifications
3.4. Prediction of Change in STAT1 Stability Due to Mutation
3.5. Pathogenicity Prediction Results
3.6. The Conservational Status and Surface Accessibility Analysis of STAT1 Protein
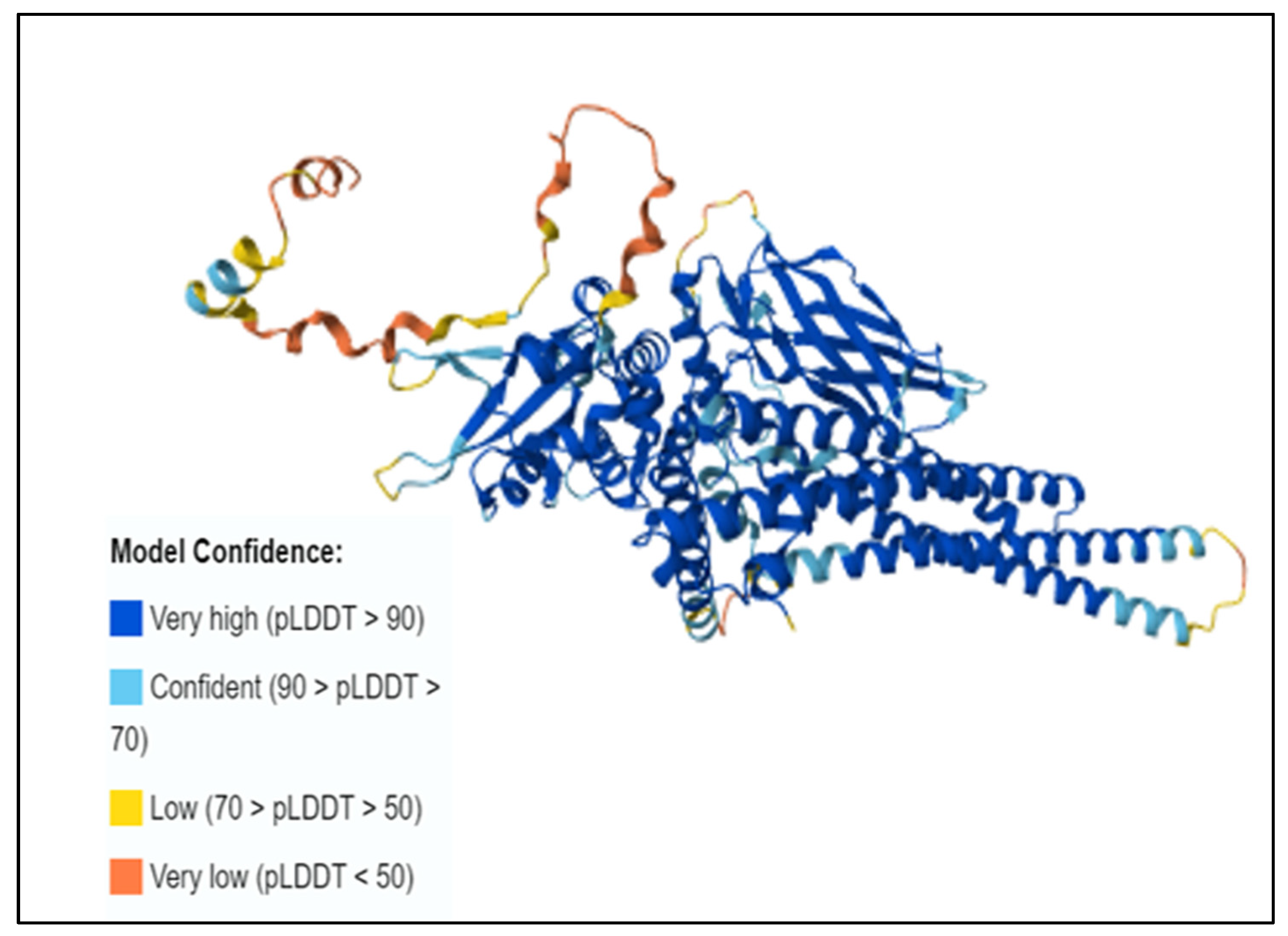
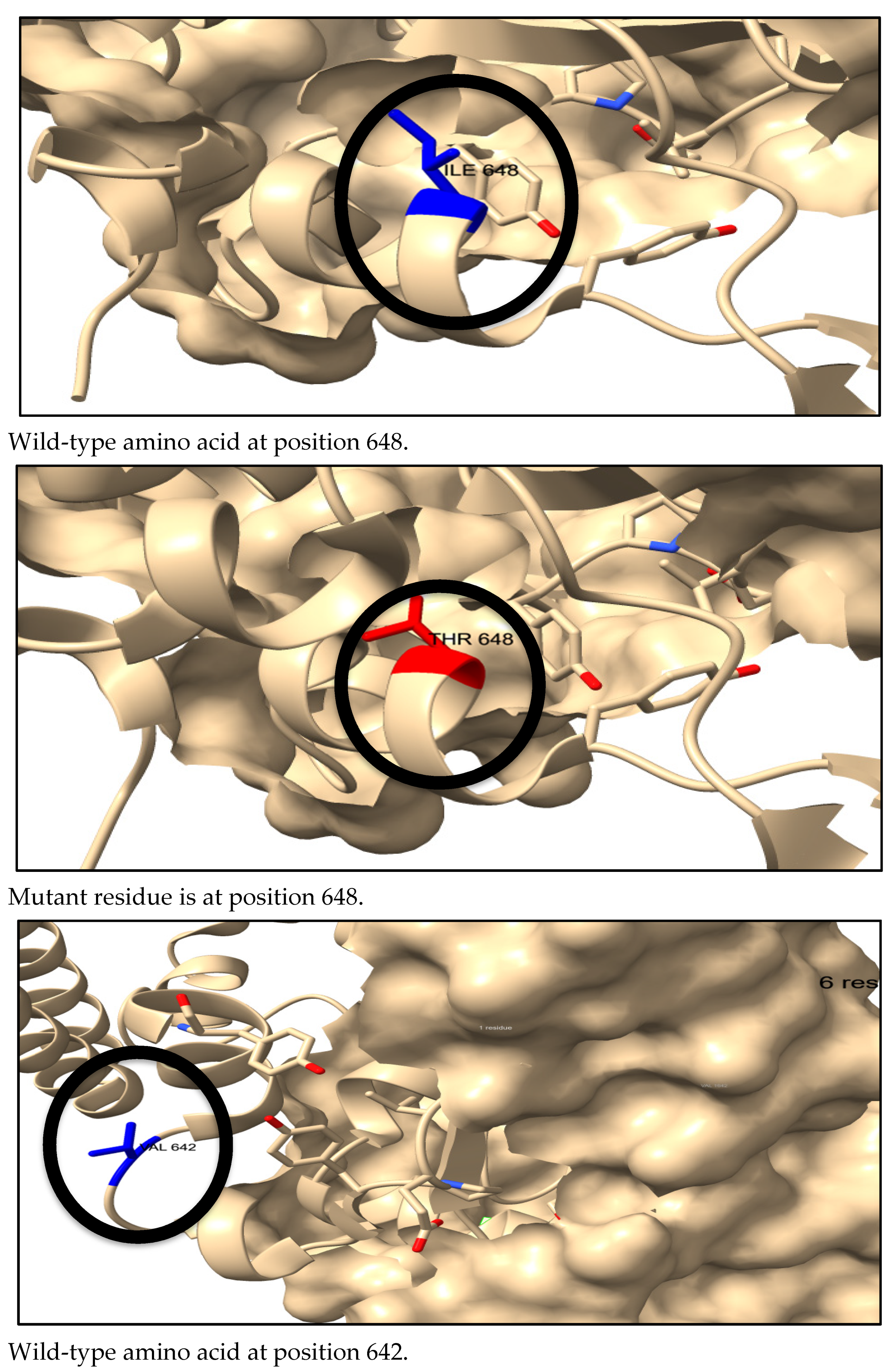
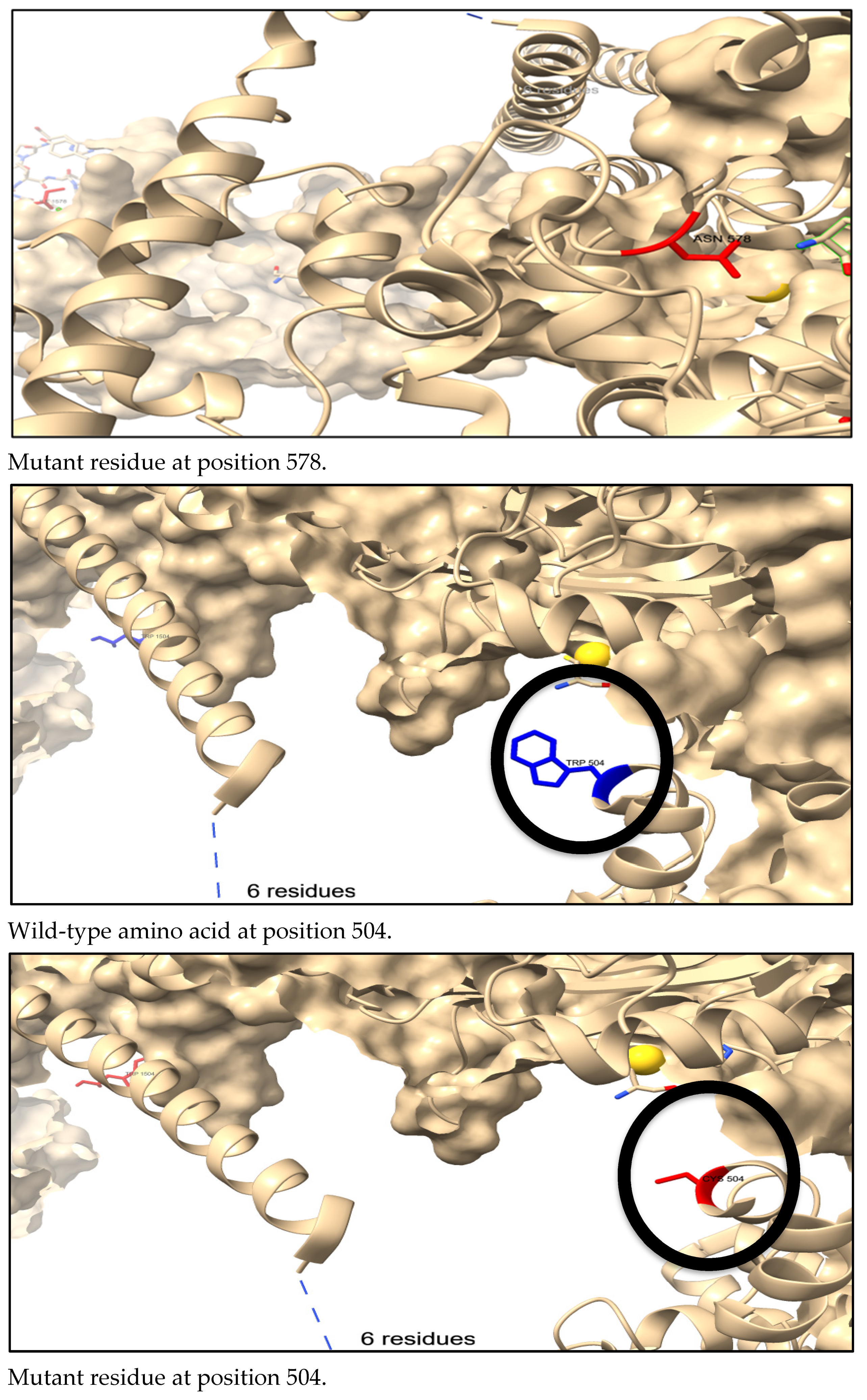
3.7. Three-Dimensional Structure Prediction by AlphaFold and SNP Visualization by ChimeraX
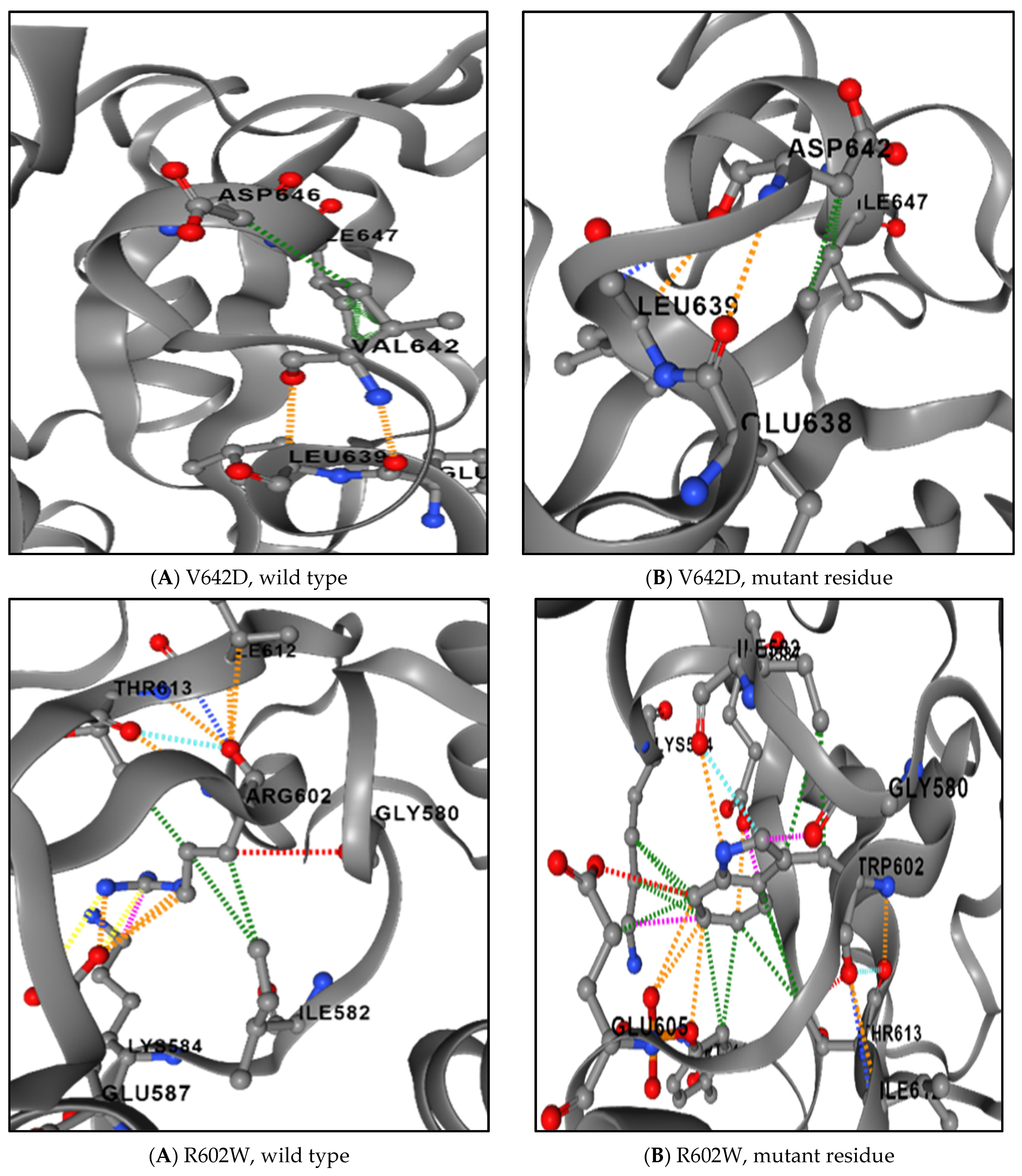
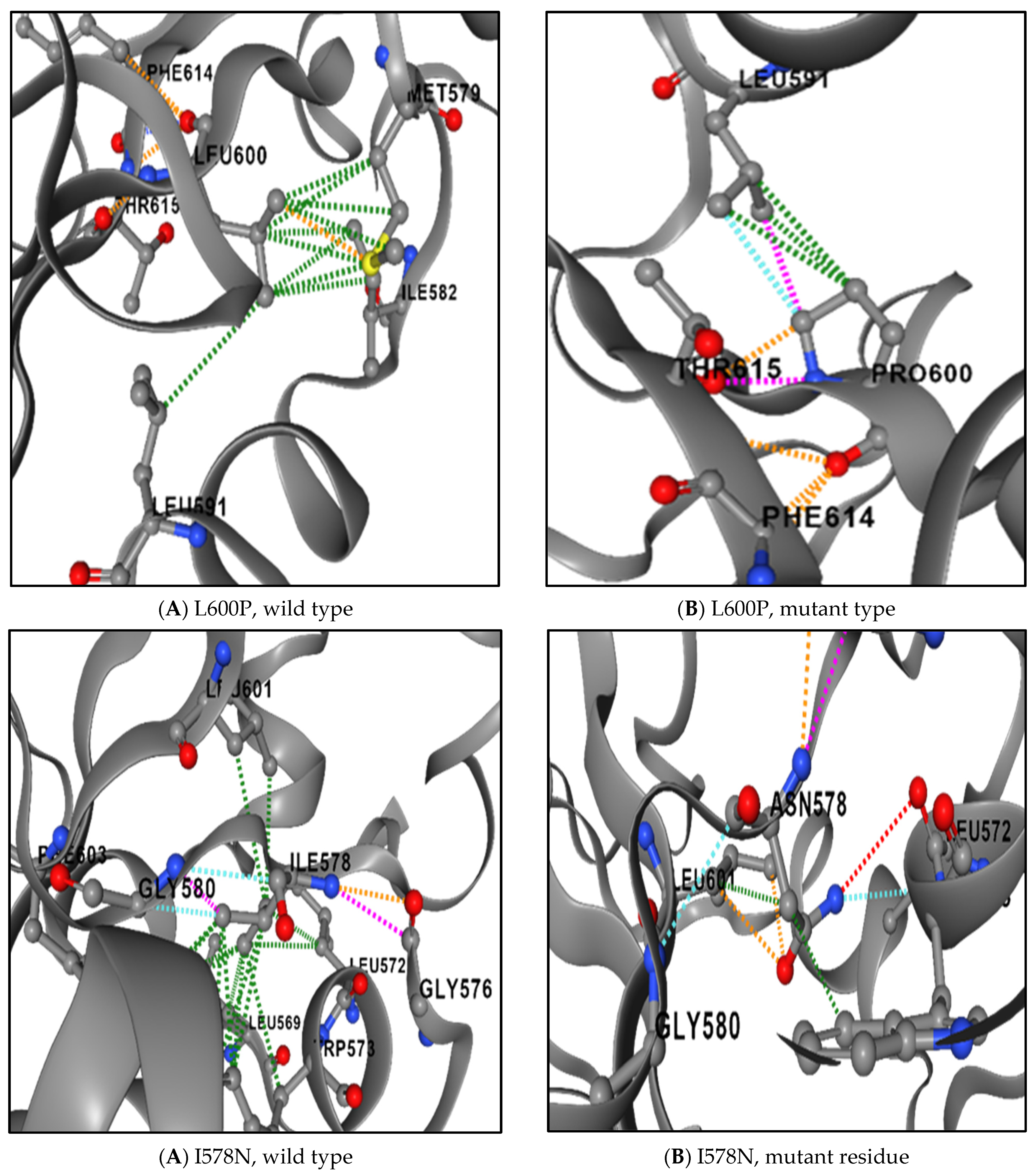
3.8. The Physical Outcome of Predicted SNPs
3.9. Domain Identification of the STAT1 Protein by the InterPro Server
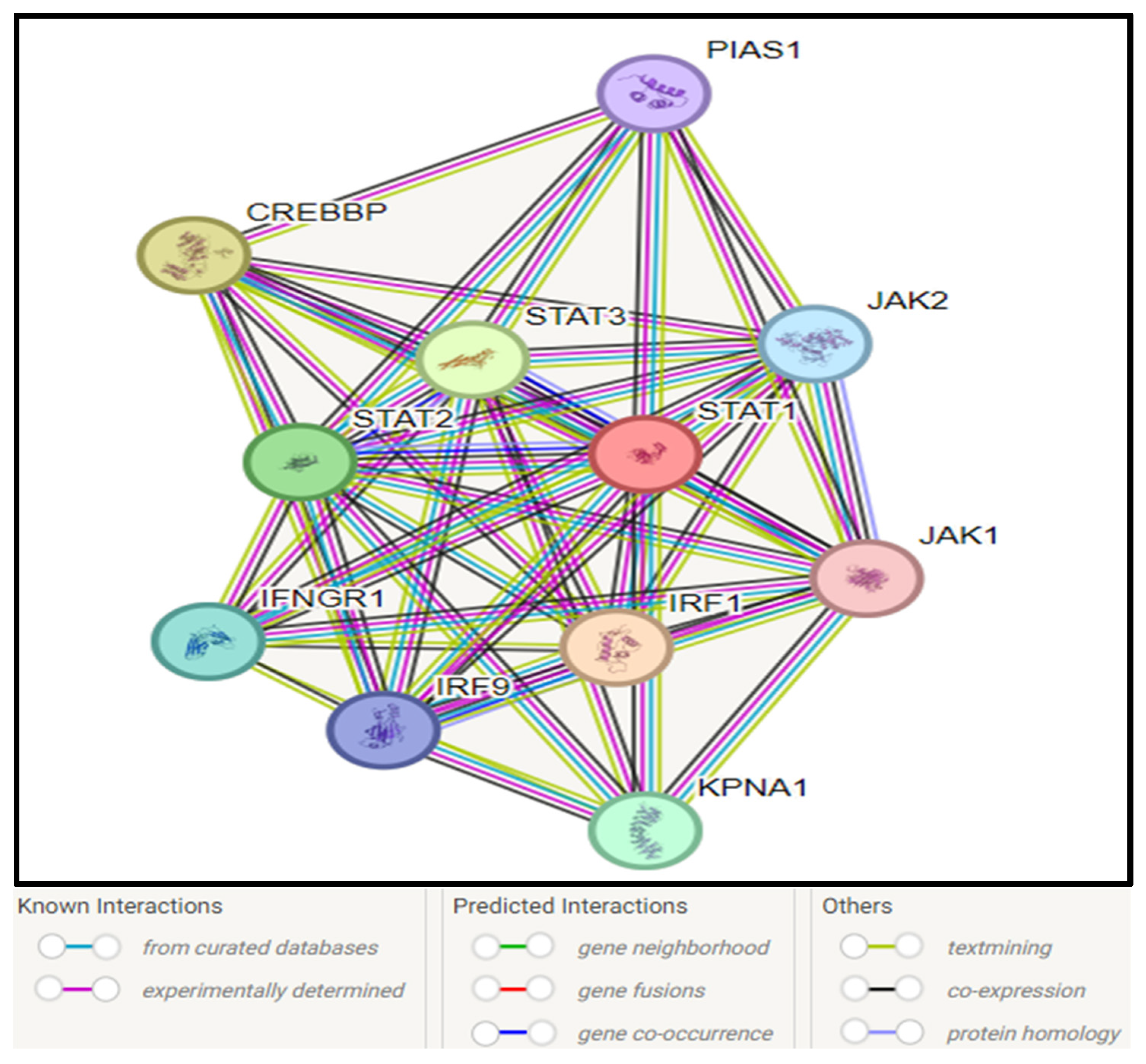
3.10. STAT1–Protein Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT proteins: A kaleidoscope of canonical and non-canonical functions in immunity and cancer. J. Hematol. Oncol. 2021, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Calò, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT proteins: From normal control of cellular events to tumorigenesis. J. Cell. Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Henriksen, M.A.; Takeuchi, K.; Schaefer, O.; Liu, B.; ten Hoeve, J.; Ren, Z.; Mao, X.; Chen, X.; Shuai, K.; et al. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 3966–3971. [Google Scholar] [CrossRef]
- Mao, X.; Ren, Z.; Parker, G.N.; Sondermann, H.; Pastorello, M.A.; Wang, W.; McMurray, J.S.; Demeler, B.; Darnell, J.E.; Chen, X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell 2005, 17, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Metwally, H.; Kishimoto, T. Distinct Phosphorylation of STAT1 Confers Distinct DNA Binding and Gene-regulatory Properties. J. Cell. Signal. 2020, 1, 50–55. [Google Scholar] [CrossRef]
- Lorenzini, T.; Dotta, L.; Giacomelli, M.; Vairo, D.; Badolato, R. STAT mutations as program switchers: Turning primary immunodeficiencies into autoimmune diseases. J. Leukoc. Biol. 2017, 101, 29–38. [Google Scholar] [CrossRef]
- Asano, T.; Utsumi, T.; Kagawa, R.; Karakawa, S.; Okada, S. Inborn errors of immunity with loss- and gain-of-function germline mutations in STAT1. Clin. Exp. Immunol. 2023, 212, 96–106. [Google Scholar] [CrossRef]
- Mizoguchi, Y.; Okada, S. Inborn errors of STAT1 immunity. Curr. Opin. Immunol. 2021, 72, 59–64. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Liongue, C.; Sobah, M.L.; Ward, A.C. Signal transducer and activator of transcription proteins at the nexus of immunodeficiency, autoimmunity and cancer. Biomedicines 2023, 12, 45. [Google Scholar] [CrossRef]
- Reich, N.C. STATs get their move on. Jak-stat 2013, 2, e27080. [Google Scholar] [CrossRef] [PubMed]
- de Prati, A.C.; Ciampa, A.R.; Cavalieri, E.; Zaffini, R.; Darra, E.; Menegazzi, M.; Suzuki, H.; Mariotto, S. STAT1 as a new molecular target of anti-inflammatory treatment. Curr. Med. Chem. 2005, 12, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cavalli, A.; Cascio, A. STAT1 and its crucial role in the control of viral infections. Int. J. Mol. Sci. 2022, 23, 4095. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Noma, K.; Mizoguchi, Y.; Karakawa, S.; Okada, S. Human STAT1 gain of function with chronic mucocutaneous candidiasis: A comprehensive review for strengthening the connection between bedside observations and laboratory research. Immunol. Rev. 2024, 322, 81–97. [Google Scholar] [CrossRef]
- Shuai, K.; Schindler, C.; Prezioso, V.R.; Darnell, J.E. Activation of transcription by IFN-γ: Tyrosine phosphorylation of a 91-kD DNA binding protein. Science 1992, 258, 1808–1812. [Google Scholar] [CrossRef]
- Heim, M.H. The Jak-STAT pathway: Cytokine signalling from the receptor to the nucleus. J. Recept. Signal Transduct. 1999, 19, 75–120. [Google Scholar] [CrossRef]
- Eilers, A.; Georgellis, D.; Klose, B.; Schindler, C.; Ziemiecki, A.; Harpur, A.G.; Wilks, A.F.; Decker, T. Differentiation-regulated serine phosphorylation of STAT1 promotes GAF activation in macrophages. Mol. Cell. Biol. 1995, 15, 3579–3586. [Google Scholar] [CrossRef]
- Decker, T.; Lew, D.J.; Mirkovitch, J.; Darnell, J.E. Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO J. 1991, 10, 927–932. [Google Scholar] [CrossRef]
- Meesilpavikkai, K.; Hirankarn, N.; Dalm, V.A.S.H.; van Hagen, P.M.; Dik, W.A.; IJspeert, H. Unraveling the Immunogenetics of STAT Proteins: Clinical Perspectives on Gain-of-Function and Loss-of-Function Variants. Asian Pac. J. Allergy Immunol. 2024, 42, 105–122. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Chen, R.; Mou, H.; Sun, G.; Yang, L.; Jia, Y.; Zhao, Q.; Wen, W.; Zhou, L.; et al. Genetic and Functional Identifying of Novel STAT1 Loss-of-Function Mutations in Patients with Diverse Clinical Phenotypes. J. Clin. Immunol. 2022, 42, 1778–1794. [Google Scholar] [CrossRef]
- Boisson-Dupuis, S.; Kong, X.-F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.-L. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Tsumura, M.; Okada, S.; Sakai, H.; Yasunaga, S.; Ohtsubo, M.; Murata, T.; Obata, H.; Yasumi, T.; Kong, X.-F.; Abhyankar, A.; et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum. Mutat. 2012, 33, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Uzel, G.; Sampaio, E.P.; Lawrence, M.G.; Hsu, A.P.; Hackett, M.; Dorsey, M.J.; Noel, R.J.; Verbsky, J.W.; Freeman, A.F.; Janssen, E.; et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J. Allergy Clin. Immunol. 2013, 131, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Hartono, S.P.; Vargas-Hernández, A.; Ponsford, M.J.; Chinn, I.K.; Jolles, S.; Wilson, K.; Forbes, L.R. Novel STAT1 Gain-of-Function Mutation Presenting as Combined Immunodeficiency. J. Clin. Immunol. 2018, 38, 753–756. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Dolan, J.G.; Forbes, L.R.; Vargas-Hernández, A.; Nishimura, S.; Okada, S.; Kersun, L.S.; Brodeur, G.M.; Heimall, J.R. Gain-of-Function STAT1 Mutation With Familial Lymphadenopathy and Hodgkin Lymphoma. Front. Pediatr. 2019, 7, 160. [Google Scholar] [CrossRef]
- Okada, S.; Asano, T.; Moriya, K.; Boisson-Dupuis, S.; Kobayashi, M.; Casanova, J.-L.; Puel, A. Human STAT1 Gain-of-Function Heterozygous Mutations: Chronic Mucocutaneous Candidiasis and Type I Interferonopathy. J. Clin. Immunol. 2020, 40, 1065–1081. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, W.; Chen, F.; Zhou, P.; Yan, Z. Chinese Pedigree of Chronic Mucocutaneous Candidiasis Due to STAT1 Gain-of-Function Mutation: A Case Study and Literature Review. Mycopathologia 2023, 188, 87–97. [Google Scholar] [CrossRef]
- Egri, N.; Esteve-Solé, A.; Deyà-Martínez, À.; Ortiz de Landazuri, I.; Vlagea, A.; García, A.P.; Cardozo, C.; Garcia-Vidal, C.; Bartolomé, C.S.; Español-Rego, M.; et al. Primary immunodeficiency and chronic mucocutaneous candidiasis: Pathophysiological, diagnostic, and therapeutic approaches. Allergol. Immunopathol. 2021, 49, 118–127. [Google Scholar] [CrossRef]
- Collins, F.S.; Brooks, L.D.; Chakravarti, A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998, 8, 1229–1231. [Google Scholar] [CrossRef]
- Arshad, S.; Ishaque, I.; Mumtaz, S.; Rashid, M.U.; Malkani, N. In-Silico Analyses of Nonsynonymous Variants in the BRCA1 Gene. Biochem. Genet. 2021, 59, 1506–1526. [Google Scholar] [CrossRef]
- Yazar, M.; Özbek, P. In Silico Tools and Approaches for the Prediction of Functional and Structural Effects of Single-Nucleotide Polymorphisms on Proteins: An Expert Review. OMICS J. Integr. Biol. 2021, 25, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S.; Almatroudi, A.; Alrumaihi, F.; Makki Almansour, N.; Aldakheel, F.M.; Rather, R.A.; Afroze, D.; Rah, B. Single nucleotide polymorphisms (SNPs) in prostate cancer: Its implications in diagnostics and therapeutics. Am. J. Transl. Res. 2021, 13, 3868–3889. [Google Scholar] [PubMed]
- Clifford, R.J.; Edmonson, M.N.; Nguyen, C.; Scherpbier, T.; Hu, Y.; Buetow, K.H. Bioinformatics tools for single nucleotide polymorphism discovery and analysis. Ann. N. Y. Acad. Sci. 2004, 1020, 101–109. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Bromberg, Y.; Yachdav, G.; Rost, B. SNAP predicts effect of mutations on protein function. Bioinformatics 2008, 24, 2397–2398. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14 (Suppl. S3), S6. [Google Scholar] [CrossRef]
- López-Ferrando, V.; Gazzo, A.; de la Cruz, X.; Orozco, M.; Gelpí, J.L. PMut: A web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017, 45, W222–W228. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Calabrese, R.; Casadio, R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics 2005, 21 (Suppl. S2), ii54–ii58. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, Q.; Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. DDMut: Predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 2023, 51, W122–W128. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Venselaar, H.; Te Beek, T.A.H.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed]
- Mulder, N.J.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Biswas, M.; Bradley, P.; Bork, P.; Bucher, P.; et al. InterPro Consortium InterPro: An integrated documentation resource for protein families, domains and functional sites. Brief Bioinform. 2002, 3, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, X.; Gao, G.; Xing, S.; Zhou, L.; Tang, X.; Zhao, X.; An, Y. Clinical Relevance of Gain- and Loss-of-Function Germline Mutations in STAT1: A Systematic Review. Front. Immunol. 2021, 12, 654406. [Google Scholar] [CrossRef]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Chapgier, A.; Boisson-Dupuis, S.; Jouanguy, E.; Vogt, G.; Feinberg, J.; Prochnicka-Chalufour, A.; Casrouge, A.; Yang, K.; Soudais, C.; Fieschi, C.; et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006, 2, e131. [Google Scholar] [CrossRef]
- Liu, L.; Okada, S.; Kong, X.-F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011, 208, 1635–1648. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Wu, W.; Wang, A.; Wan, Z.; van de Veerdonk, F.L.; Li, R. New and recurrent STAT1 mutations in seven Chinese patients with chronic mucocutaneous candidiasis. Int. J. Dermatol. 2017, 56, e30–e33. [Google Scholar] [CrossRef]
- Breuer, O.; Daum, H.; Cohen-Cymberknoh, M.; Unger, S.; Shoseyov, D.; Stepensky, P.; Keller, B.; Warnatz, K.; Kerem, E. Autosomal dominant gain of function STAT1 mutation and severe bronchiectasis. Respir. Med. 2017, 126, 39–45. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Amino Acid Change | SIFT | Poly-Phen-2 | Provean | SNAP2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Prediction | TI | Effect | Score | Effect | Score | Prediction | Score | |||
| 1 | rs1173266737 | P728A | Deleterious | 0 | PB | 0.974 | Deleterious | −3.14 | E | 15 |
| 2 | rs1374373369 | D674V | Deleterious | 0.01 | PB | 0.989 | Deleterious | −7.327 | E | 47 |
| 3 | rs771679419 | Y668F | Deleterious | 0 | PS | 0.609 | Deleterious | −3.583 | E | 67 |
| 4 | rs759271255 | I648T | Deleterious | 0.01 | PB | 1 | Deleterious | −4.197 | E | 56 |
| 5 | rs752542806 | V642D | Deleterious | 0 | PB | 0.994 | Deleterious | −5.149 | E | 60 |
| 6 | rs1387961263 | L639F | Deleterious | 0 | PB | 1 | Deleterious | −3.489 | E | 7 |
| 7 | rs1209841496 | R602W | Deleterious | 0 | PB | 1 | Deleterious | −7.4 | E | 87 |
| 8 | rs137852678 | L600P | Deleterious | 0 | PB | 1 | Deleterious | −6.472 | E | 91 |
| 9 | rs1398307167 | P596Q | Deleterious | 0.01 | PS | 0.951 | Deleterious | −3.974 | E | 57 |
| 10 | rs1398307167 | P596L | Deleterious | 0.01 | PB | 0.966 | Deleterious | −5.853 | E | 58 |
| 11 | rs767475430 | I578N | Deleterious | 0 | PB | 0.1 | Deleterious | −6.392 | E | 80 |
| 12 | rs113988352 | I561T | Deleterious | 0 | PB | 0.981 | Deleterious | −4.143 | E | 38 |
| 13 | rs1803838 | P538L | Deleterious | 0.03 | PS | 0.588 | Deleterious | −3.097 | E | 10 |
| 14 | rs916580554 | W504C | Deleterious | 0 | PB | 0.999 | Deleterious | −11,937 | E | 48 |
| 15 | rs1185249247 | S503N | Deleterious | 0 | PS | 0.949 | Deleterious | −2.621 | E | 50 |
| 16 | rs866554932 | P481R | Deleterious | 0.04 | PB | 0.1 | Deleterious | −6.451 | E | 6 |
| 17 | rs935654762 | V455A | Deleterious | 0 | PB | 0.997 | Deleterious | −3.255 | E | 35 |
| 18 | rs527393923 | T450M | Deleterious | 0 | PB | 0.1 | Deleterious | −4.499 | E | 56 |
| 19 | rs760409880 | L448F | Deleterious | 0 | PS | 0.816 | Deleterious | −3.135 | E | 33 |
| 20 | rs776192196 | P326L | Deleterious | 0 | PB | 0.996 | Deleterious | −6.947 | E | 29 |
| 21 | rs763976174 | R304H | Deleterious | 0.02 | PS | 0.850 | Deleterious | −2.809 | E | 52 |
| 22 | rs751403509 | R304C | Deleterious | 0 | PB | 0.1 | Deleterious | −4.767 | E | 39 |
| 23 | rs779371351 | I248N | Deleterious | 0 | PB | 0.1 | Deleterious | −5.877 | E | 38 |
| 24 | rs779371351 | I248T | Deleterious | 0 | PB | 0.1 | Deleterious | −4.218 | E | 42 |
| 25 | rs1017740241 | C247Y | Deleterious | 0 | PB | 0.1 | Deleterious | −9.192 | E | 49 |
| 26 | rs763588438 | V149G | Deleterious | 0.01 | PB | 0.987 | Deleterious | −4.942 | E | 48 |
| 27 | rs1482374494 | A119T | Deleterious | 0 | PB | 0.1 | Deleterious | −3.113 | E | 26 |
| 28 | rs756147217 | P98S | Deleterious | 0 | PB | 0.1 | Deleterious | −5.885 | E | 48 |
| 29 | rs865962653 | S51L | Deleterious | 0.01 | PS | 0.883 | Deleterious | −4.4 | E | 34 |
| 30 | rs781389511 | A46T | Deleterious | 0 | PB | 0.1 | Deleterious | −2.543 | E | 1 |
| 31 | rs34255470 | I30T | Deleterious | 0.02 | PB | 0.1 | Deleterious | −3.391 | E | 17 |
| 32 | rs11549696 | P27T | Deleterious | 0 | PB | 0.1 | Deleterious | −6.503 | E | 64 |
| 33 | rs1233778383 | W4C | Deleterious | 0 | PB | 1 | Deleterious | −10.563 | E | 23 |
| nsSNP | Amino Acid Change | P MUT | PhD-SNP | SNP&GO | PANTHER | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Prediction | Score | Prediction | RI | Prediction | RI | Effect | Preservation Time | |||
| 1 | rs1374373369 | D674V | Disease | 90% | Disease | 9 | Disease | 8 | probably damaging | 1036 |
| 2 | rs759271255 | I648T | Disease | 92% | Disease | 6 | Disease | 6 | probably damaging | 842 |
| 3 | rs752542806 | V642D | Disease | 87% | Disease | 5 | Disease | 8 | probably damaging | 455 |
| 4 | rs1209841496 | R602W | Disease | 93% | Disease | 8 | Disease | 3 | probably damaging | 1237 |
| 5 | rs137852678 | L600P | Disease | 93% | Disease | 9 | Disease | 8 | probably damaging | 1036 |
| 6 | rs767475430 | I578N | Disease | 93% | Disease | 8 | Disease | 6 | probably damaging | 1237 |
| 7 | rs916580554 | W504C | Disease | 91% | Disease | 6 | Disease | 8 | probably damaging | 750 |
| 8 | rs527393923 | T450M | Disease | 86% | Disease | 1 | Disease | 2 | probably damaging | 1036 |
| 9 | rs865962653 | S51L | Disease | 88% | Disease | 5 | Disease | 1 | probably damaging | 750 |
| SNP ID | Amino Acid Change | MutPred 2 Score | Affected PROSITE and ELM Motifs | Molecular Mechanisms | Probability | p-Value | |
|---|---|---|---|---|---|---|---|
| 1 | rs1374373369 | D674V | 0.813 | Gain of Strand Gain of Acetylation at K673 | 0.26 0.25 | 0.04 0.01 | |
| 2 | rs759271255 | I648T | 0.893 | Altered Stability | 0.16 | 0.02 | |
| 3 | rs752542806 | V642D | 0.867 | ELME000063 ELME000085 ELME000147 ELME000155 ELME000220 ELME000233 | Altered Ordered interface | 0.35 | 4.2 × 10−3 |
| Gain of Relative solvent Accessibility | 0.30 | 7.3 × 10−3 | |||||
| Altered Transmembrane protein | 0.18 | 8.6 × 10−3 | |||||
| Altered DNA binding | 0.15 | 0.04 | |||||
| 4 | rs1209841496 | R602W | 0.896 | - ELME000328 ELME000052 ELME000062 | Gain of Strand | 0.27 | 0.02 |
| Altered Stability | 0.09 | 0.05 | |||||
| 5 | rs137852678 | L600P | 0.965 | ELME000052 ELME000328 | Gain of Intrinsic disorder | 0.31 | 0.04 |
| Altered Stability | 0.28 | 6.6 × 10−3 | |||||
| 6 | rs767475430 | I578N | 0.936 | PS00008 | |||
| 7 | rs916580554 | W504C | 0.807 | ELME000197 | |||
| 8 | rs527393923 | T450M | 0.373 | ||||
| 9 | rs865962653 | S51L | 0.665 | ELME000063 ELME000147 ELME000336 | Altered transmembrane protein | 0.23 | 2.4 × 10−3 |
| SNP ID | Amino Acid Change | I Mutant 3 | MUpro | DDMUT | |||||
|---|---|---|---|---|---|---|---|---|---|
| Stability | RI | DDG (kcal/mol) | Stability | DDG (kcal/mol) | Stability | DDG (kcal/mol) | |||
| 1 | rs759271255 | I648T | Decrease | 9 | −2.43 | Decrease | −2.4802937 | Destabilizing | −2.93 |
| 2 | rs752542806 | V642D | Decrease | 8 | −1.85 | Decrease | −1.8071037 | Destabilizing | −1.11 |
| 3 | rs1209841496 | R602W | Decrease | 3 | −0.20 | Decrease | −1.0486884 | Destabilizing | −0.19 |
| 4 | rs137852678 | L600P | Decrease | 3 | −1.54 | Decrease | −1.6074419 | Destabilizing | −3.06 |
| 5 | rs767475430 | I578N | Decrease | 5 | −1.92 | Decrease | −0.98144877 | Destabilizing | −0.84 |
| 6 | rs916580554 | W504C | Decrease | 8 | −1.41 | Decrease | −0.86533645 | Destabilizing | −0.73 |
| SNP ID | Substitution | Alpha-Missense Pathogenicity | Alpha-Missense Prediction | |
|---|---|---|---|---|
| 1 | rs759271255 | I648T | 0.9875 | Likely Pathogenic |
| 2 | rs752542806 | V642D | 0.9916 | Likely Pathogenic |
| 3 | rs1209841496 | R602W | 0.9982 | Likely Pathogenic |
| 4 | rs137852678 | L600P | 0.9998 | Likely Pathogenic |
| 5 | rs767475430 | I578N | 0.9986 | Likely Pathogenic |
| 6 | rs916580554 | W504C | 0.9815 | Likely Pathogenic |
| SNP ID | Amino Acid Change | Conservation Score | Prediction | |
|---|---|---|---|---|
| 1 | rs759271255 | I648T | 8 | Conserved and buried |
| 2 | rs752542806 | V642D | 6 | Buried |
| 3 | rs120984149 | R602W | 9 | (functional residues), highly conserved and exposed |
| 4 | rs137852678 | L600P | 9 | (structural residues), highly conserved and buried |
| 5 | rs767475430 | I578N | 9 | (structural residues), highly conserved and buried |
| SNPs | Difference in Size | Difference in Charge | Difference in Hydrophobicity | Disrupt Hydrogen Bond | Affect Contact with Ligand Molecules | |
|---|---|---|---|---|---|---|
| 1 | I648T | Yes | No | Yes | No | Yes |
| 2 | V642D | Yes | No | Yes | No | Yes |
| 3 | R602W | Yes | No | Yes | No | Yes |
| 4 | L600P | Yes | No | Yes | No | No |
| 5 | I578N | Yes | No | Yes | Yes | Yes |
| 6 | W504C | Yes | No | Yes | No | Yes |
| STAT1 Domains (Position) | SNPs |
|---|---|
| STAT1, SH2 domain (557–707) | Y668F, I648T, V642D, R602W, and L600P |
| STAT1 transcription factor, DNA binding domain (323–458) | R304C |
| SH2 domain (578–638) | I578N |
| Src homology 2 (SH2) domain profile (573–670) | I578N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamal, E.; Kaddam, L.A.; Ahmed, M.; Alabdulkarim, A. Integrating Artificial Intelligence and Bioinformatics Methods to Identify Disruptive STAT1 Variants Impacting Protein Stability and Function. Genes 2025, 16, 303. https://doi.org/10.3390/genes16030303
Kamal E, Kaddam LA, Ahmed M, Alabdulkarim A. Integrating Artificial Intelligence and Bioinformatics Methods to Identify Disruptive STAT1 Variants Impacting Protein Stability and Function. Genes. 2025; 16(3):303. https://doi.org/10.3390/genes16030303
Chicago/Turabian StyleKamal, Ebtihal, Lamis A. Kaddam, Mehad Ahmed, and Abdulaziz Alabdulkarim. 2025. "Integrating Artificial Intelligence and Bioinformatics Methods to Identify Disruptive STAT1 Variants Impacting Protein Stability and Function" Genes 16, no. 3: 303. https://doi.org/10.3390/genes16030303
APA StyleKamal, E., Kaddam, L. A., Ahmed, M., & Alabdulkarim, A. (2025). Integrating Artificial Intelligence and Bioinformatics Methods to Identify Disruptive STAT1 Variants Impacting Protein Stability and Function. Genes, 16(3), 303. https://doi.org/10.3390/genes16030303