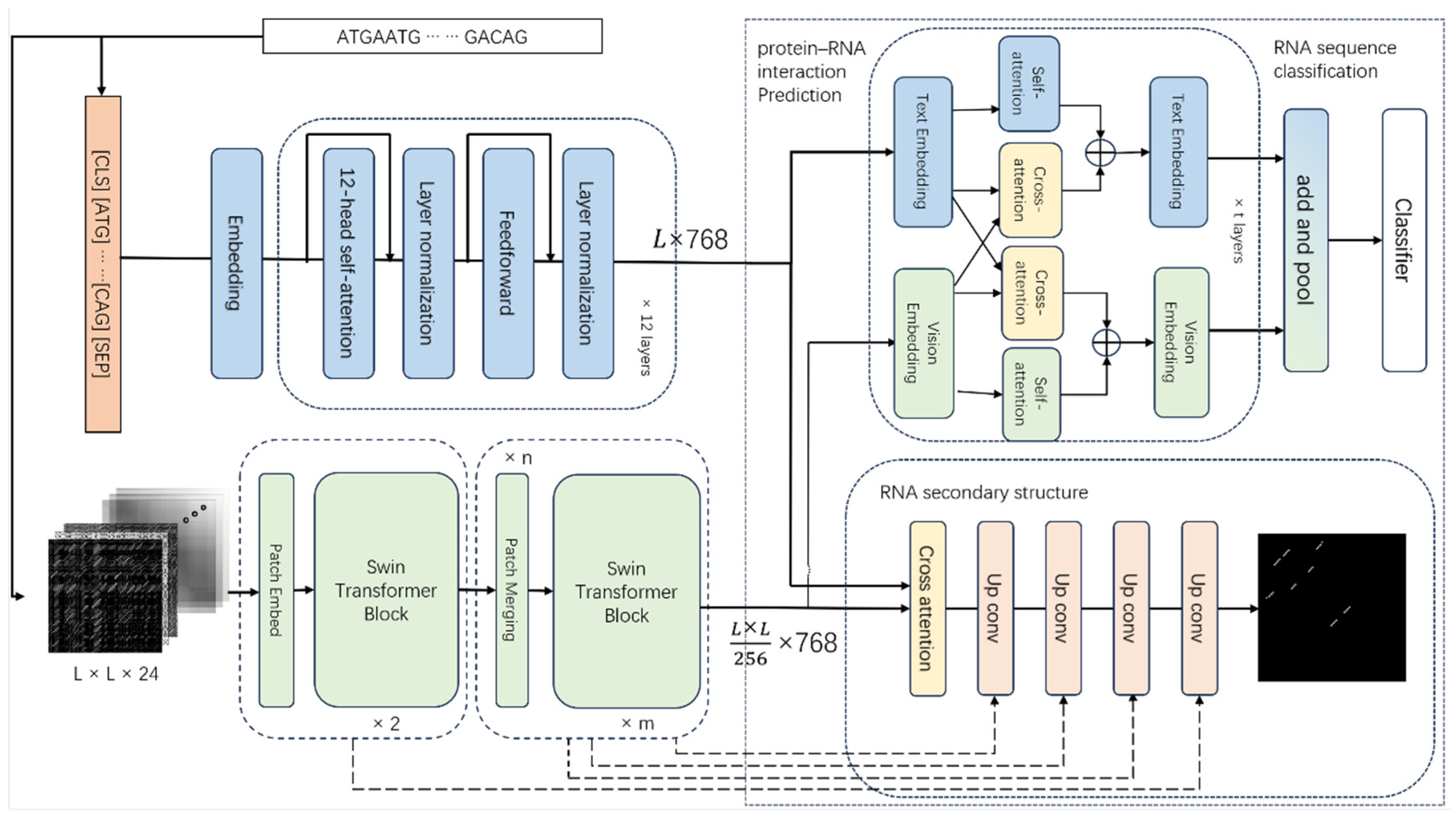
1. Introduction
RNA is a pivotal molecule in the central dogma of molecular biology. As a multi-functional macromolecule and genetic information carrier, it describes the flow of genetic information from DNA to RNA and ultimately to protein [
1]. RNA plays a crucial role in various cellular processes, such as gene expression, regulation, and catalysis, and has become an essential regulatory factor influencing life processes [
2]. Given RNA’s significance in biological systems, there is an increasing demand for efficient and accurate methods to analyze RNA sequences.
Traditionally, RNA sequences are analyzed through experimental techniques such as RNA sequencing and microarray analysis [
3]. For instance, cDNA preparation, sequencing and read mapping on the genome and across splices [
4], and the widely adopted dUTP method [
5]. However, these methods are often time-consuming and expensive. In recent years, computational approaches based on machine learning have been employed to analyze RNA sequences. For example, Adam McDermaid et al. proposed GeneQC [
6], a machine learning tool, to accurately estimate the reliability of each gene expression level derived from RNA-Seq datasets. With the advent of artificial intelligence, deep learning methods have also been utilized for RNA sequence analysis and various downstream tasks. These methods have been widely applied in specific tasks, for example, protein–RNA interaction prediction by DeepBind [
7] and RNA secondary structure prediction by MXFold2 [
8].
Recently, pre-trained language models have achieved remarkable success in various natural language processing (NLP) tasks, such as text classification, question answering, and language translation. Researchers have increasingly applied the pre-trained language models like BERT to simulate and analyze the large-scale biological sequences. For instance, in RNAErnie proposed by Wang et al. [
9], RNA motifs are incorporated as biological priors. This approach introduces the motif-level random masking and sub-sequence-level masking in language modeling to enhance the pre-training. Additionally, RNA types are added as the stop words during pre-training, which can effectively separate the word embeddings among different RNA types, leading to the improved representational capacity. In DNABERT proposed by Ji et al. [
10], biological sequences are tokenized using k-mers, followed by BERT-based pre-training, which can effectively capture sequence information. Followingly, Zhou et al. [
11] introduced an advanced approach, DNABERT-2, to replace the k-mer tokenization with Byte Pair Encoding (BPE), overcoming the limitations of k-mer tokenization. This enhanced tokenization strategy, combined with BERT-based pre-training, significantly improves the model’s learning capability.
Currently, most benchmark models use sequences as input, relying solely on the unimodal representation of sequences to predict RNA structures and perform various downstream tasks. However, as we know for RNA, the biological sequence determines its structure, thereby affecting its function. Consequently, relying solely on the sequence-based unimodal benchmark models may not yield the accurate results for downstream analysis. In the traditional deep learning field, structural information can be used to complement sequence information for specific tasks. For instance, the protein–RNA interaction prediction, PrismNet, proposed by Xu et al. [
12], incorporates icSHAPE data (which represents single-stranded and double-stranded structural information of sequences) as a supplement, achieving significant prediction improvement. Therefore, it is crucial to explore multimodal approaches that integrate sequence and structural information to construct benchmark models, thereby enhancing the analysis of downstream tasks.
In terms of structural information, we notice that there is the emergence of some vision-based deep learning models for RNA downstream analysis. For example, in the RNA secondary structure prediction (RSS), UFold [
13] is proposed to extract features based on RNA secondary structure information, while RNAformer [
14] is designed, which uses the word embeddings to extract features. In this work, we firstly construct novel structural vision features based on RNA secondary structure matching rules. Using these features, we pre-train a vision-based benchmark model SWIN-RNA with the SWIN transformer [
15], and then integrate the vision benchmark model with a sequence benchmark model to form a multimodal model DRFormer. We conduct extensive downstream analyses by this multimodal model, including the sequence classification, protein–RNA interaction prediction, and RNA secondary structure prediction. Additionally, we perform the generalization experiment on DNA downstream tasks. All experiments yield satisfactory results, demonstrating the versatility and effectiveness of DRFormer as a general-purpose solution.
3. Results
3.1. Evaluation Metrics
Taking into account the imbalance of data distribution and the difference of tasks, we employ different evaluation indicators for various tasks. In the RSS task, we apply F1 as the main indicator, and Sensitivity (Sen) and Positive Predictive Value (PPV) as auxiliary indicators. Here, the F1 value is the harmonic mean of precision and recall, which is used to comprehensively evaluate the performance of classification model. PPV, Sen, and F1 are calculated as follows:
where TP represents the true positive examples, FP represents the false positive examples, and FN represents the false negative examples.
Other involved evaluation indicators include MCC and ACC. MCC is an indicator used to measure the model performance in a binary classification problem, and its value ranges from −1 to 1. ACC (Accuracy) is the ratio of correctly classified samples to the total number of samples.
In addition, we use AUPRC curves (precision–recall curve) and AUROC (receiver operating characteristic curve) to evaluate the performance of classification models. In the protein–RNA interaction prediction, RNA sequence classification, and DNA downstream tasks, MCC is referred as the primary metric and AUC as the secondary metric for evaluation.
3.2. Feature Ablation
We first conduct an ablation experiments on the selected features, and confirm the effectiveness of these features by comparing them on the RSS task. The features include the base pairing feature (BPS), the structural feature (SS), and the distance feature (DIS). We first compare the base pairing feature and structural feature separately, then combine the base pairing feature with structural feature for comparison, and finally add the distance feature for comparison. We compare the results on the TS0_112 dataset, which are shown in
Table 1. The results of F1 are 0.587 and 0.613 by the base pairing feature and structural feature, respectively. When the base pairing and structural features are combined together, the F1 changes to 0.631. After adding the distance feature, the F1 reaches 0.639, which is 8.9% higher than using only the base pairing feature and 4.2% higher than using only the structural feature.
Since the structural features include four separate features, namely the longest possible secondary structure (LPSS), RNA coding matrix (RCM), sequence repeat structure (SRS), and base missing structure (BMS), we use the base pairing signature (BPS) as the baseline feature, and then add the corresponding structural features for comparison. The comparison results on the TS0_112 dataset are shown in
Table 2. It is found that the F1 of adding any separate structural feature is higher than that of using the base pairing feature alone, and lower than that of the combination of the base pairing feature and four structural features. This shows that our structural features can well compensate for the reduced performance caused by the sparsity of the base pairing feature.
To explore the generalization performance of these features, we also test them on the TestSetA_112 and TestSetB_112 datasets. We find that these base pairing signature (BPS) and structural signature (SS) features have good generalization results. The F1 index by the base pairing feature is generalized from 0.790 in TestSetA_112 to 0.328 in TestSetB_112, while the result by the structural feature is changed from 0.786 in TestSetA_112 to 0.321 in TestSetB_112, with similar generalization capabilities. Additionally, the combination of these two features achieves better generalization performance, from 0.816 to 0.358 in the F1 index, with a relatively 9.1% improvement on the base pairing feature alone and a relatively 11.5% improvement on the structural feature alone on TestSetB_112. The use of all features has a relatively 12.8% improvement in generalization performance compared to the base pairing signature.
3.3. SWIN-RNA Pre-Training Ablation
We conduct the ablation experiment on random weights and pre-trained weights of SWIN-RNA on the RSS prediction and RNA classification tasks, and compare the performance by fine-tuning two kinds of weights, as shown in
Table 3. We find that the result by pre-trained weights surpasses that by random weights in all indicators. In addition, we visualize the feature embedding result on the nRC dataset under two weights and the feature embedding after fine-tuning the pre-trained weights, as shown in
Figure 2. We find that the feature embedding of different classes presents a large random distribution by the random initialization, while a few categories have been clustered together through the pre-trained weights, such as ribozyme, 5S_rRNA, and 5_8S_rRNA.
In addition, we plot the local average entropy and local average attention distance of the attention heads at different layers in SWIN-RNA, as shown in
Figure 3. We compare the results using weights by the pre-trained model and random initialization.
The average entropy by the pre-trained model is smaller than that of the random initialization both locally and globally, indicating that the attention area is more focused (smaller entropy), and the attention of the pre-trained model is more local (smaller average distance). Namely, SWIN-RNA has the lower attention distance and entropy compared with the random initialization. At the same time, we notice that the distance and entropy of different attention heads are distributed in a large range, which can pay attention to different features and have a larger attention range, making the model have a more focus on local and global tokens, with a wide and concentrated accumulation.
3.4. Multimodal Ablation
We conduct the ablation experiment on the single sequence model, single vision model and their combined multimodality. In the protein–RNA interaction prediction task, we select several datasets (CPSF3 in the HEK293 lineage, CDC40 in the HepG2 lineage, and ATXN2 in the HEK293T lineage), in which there are poor prediction results by the PrismNet model. By applying our DRFormer, the prediction performance improves significantly compared to using either a single sequence modality or a vision modality alone, as shown in
Table 4. At the same time, we find that there are different performances by DRFormer with different numbers of multimodal fusion block layers on these datasets. Overall, a DRFormer model with two multimodal fusion block layers performs the optimized prediction, and for most evaluation indexes, DRFormer outperforms PrismNet and DNABERT methods. Especially, in terms of the F1 index on the CDC40_HepG2 dataset, SWIN-RNA has been better than DNABERT, indicating that our pre-training model can effectively extract features.
3.5. RNA Downstream Task Analysis
3.5.1. RSS Prediction
For the RSS prediction, we select UFold [
13] (model based on image vision), RNAErnie [
9] (a relatively new large model), MXfold2 [
8] (a method based on thermodynamics), and SPOT-RNA [
26] (transfer learning–based model) for the comparison. The comparison results are shown in
Table 5.
On the TS0_112 dataset, DRFormer outperforms above four models. Note that SPOT-RNA trains on the complete TR0 and PDB datasets, and then uses a multimodal fusion method for prediction, so it achieves the relatively good results on the corresponding TS0 subset. However, our DRFormer is only trained on TR0_112 with less training data, and achieves better results than SPOT-RNA, which shows that our model can effectively capture the corresponding structural information. In addition, it is found that our model also achieves the best results in F1 and precision on the TS0_112 dataset, leading the second SPOT-RNA by about 1% in F1 and about 10.8% in precision.
For SPOT-RNA and DRFormer, we also visualize some portions of predicted structures for analysis in
Figure 4 (
Appendix B.1 Figure A1 for more results). In
Figure 4a, our model predicts more correct structures than SPOT-RNA (the green box is the correct region, and our model predicted four regions), which shows that our predicted structures are more accurate than SPOT-RNA. Although SPOT-RNA tries to predict some other structures, most of them are wrong as shown in the red boxes. In
Figure 4b, our model only predicts one incorrect structure, while the prediction results of SPOT-RNA have a relatively higher error rate, and SPOT-RNA only predicts one structure correctly, which shows that the structure predicted by our model is more credible.
In addition, we do the motif analysis of subsequences existing paired relationship on the TS0_112 dataset, as shown in
Figure 5. In this figure, the left shows the motif with paired bases in the real structure, and the right shows the motif with paired bases by our prediction model. We find that the predicted motif result is almost same the real result, which shows that our prediction model can precisely obtain the structural information.
3.5.2. RNA Sequence Classification
In this section, we evaluate our model for the RNA sequence classification, and choose the commonly used RNAcon [
27], nRC [
21], ncRFP [
28], RNAGCN [
29], ncRDeep [
30], and several large models such as RNABERT [
31], RNAErnie [
9], and RNA-MSM [
32] for comparison.
The comparison results are shown in the left of
Figure 6 (
Appendix B.2 Table A1). In the RNA sequence classification, our DRFormer achieves the best results compared with above comparative models, followed by RNAErnie. Note that RNAErnie has added RNA type as a special IND label (a special token like CLS token) for pre-training, and it achieves good results when performing RNA sequence classification on the nRC dataset. Our multimodal model DRFormer effectively combines sequence and structural information to obtain the best result of 0.948, which is about 1.12% ahead of the second RNAErnie. The right of
Figure 6 shows the classification confusion matrix of our model. We can see that our model can make satisfactory predictions for most categories of RNA sequences, such as 5S_rRNA, Intron_gpll, tRNA, etc., with an accuracy rate of more than 95%.
3.5.3. Protein–RNA Interaction Prediction
In the protein–RNA interaction prediction, we select PrismNet and PrismNet_Str [
12], GraphProt [
33], BERT-RBP [
34], and RNAErnie [
9] for comparison. The comparison results are shown in
Figure 7 and
Figure 8 (
Appendix B.3 Table A2). We observe that PrismNet_Str introduces additional icSHAPE structural information compared to PrismNet, and it indeed brings a significant improvement, indicating that structural feature can provide useful information for prediction. Of course, as the first large model proposed in the protein–RNA interaction prediction task, BERT-RBP shows a good indicator of MCC, far exceeding those machine learning models and non-large deep learning models. Note that the MCC of RNAErnie on some RBPs (RNA-binding proteins) is always 0, which may be influenced by the instability of the model. At the same time, the performance of RNAErnie is not as good as BERT-RBP. Although RNAErnie is leading in the F1 index, it may be the result of RNAErnie oscillation, as the MCC for the remaining is 0 two times.
Our multimodal model applies a variety of structural feature and also uses a sequence module to combine sequence information. Therefore, it is better than the second-top BERT-RBP model based on the pre-trained DNABERT in terms of MCC, AUC, F1, and other indicators. And compared with PrismNet, PrismNet_Str, GraphProt, and RNAErnie, our model is significantly in the lead in most RBP performance indices.
In addition, we visualize the attention analysis and the actual structure distribution on the RNA sequence as the case study in
Figure 9. It illustrates a total of three RNA sequence fragments, in which A and B are different RNA fragments corresponding to SND1 in the K562 lineage, and C is the RNA fragment corresponding to AUH in the K562 lineage. Through the analysis of the first and second fragments, we find that our model does not express well (the green box part) on the single-stranded structure (icSHAPE score tends to 1), and would prefer double-stranded structures (icSHAPE score tends to 0). This is consistent with what we know about the preference of SND1, which prefers double-stranded structures [
12].
At the same time, we also analyze some motifs in the mCross database [
35]. As shown in
Figure 9D, the motif of SND1 in the K562 lineage has a certain significance in the GGCC sequence segment (red box in
Figure 9A,B).
Figure 9E shows the motif of AUH in the K562 lineage. It is also significant in UAUC, which is consistent with our high attention expression area (red box in
Figure 9C), indicating that our model can pay good attention to the corresponding sequence segment.
3.6. Generalizability Analysis—DNA Downstream Task Analysis
In order to verify the generalization performance of our DRFormer model, we also analyze its applicability on the DNA downstream tasks. We conduct the experiment on some parts of GUE datasets. Specifically, we refer to the 13 datasets of Zhou et al. [
9] and analyze three tasks of Transcription Factor Prediction (Human), Core Promoter Detection (Human), and Transcription Factor Prediction (Mouse). At the same time, we compare it with the current large models based on sequence text, such as DNABERT, Nucleotide Transformer (NT) [
36], and RNAErnie [
9]. We find that our model performs better on most datasets, which is shown in
Figure 10 (
Appendix B.4 Table A3,
Table A4 and
Table A5).
For the long sequences, we first adopt the following strategy for treatment: we choose the first 112 bases of the original sequence for model training and prediction, and then select 13 datasets from the remaining GUE dataset [
9] and compare two tasks of Epigenetic Marks Prediction (EMP) and Promoter Detection (PD) for analysis. The overall performance is shown in
Table 6 (
Appendix B.4 Table A6 for detailed results). We find that DRFormer leads in 11 of 13 datasets, with an average score of 0.601, 4.34% ahead of the second method. This shows that even if our model only uses part of the sequence for training, our model still works well. Of course, for the complete long sequences, we can also employ the complete sequence for training and prediction. We conduct the experiment for PD task on the tata dataset. After training through the complete sequence, we find our MCC is 0.798, which is about 5.7% higher than the sequence truncation strategy.
4. Discussion
This paper proposes a novel benchmark model DRFormer for the analysis of various downstream tasks of RNA sequences. The main contributions of this model are as follows: (1) in terms of structural feature extraction, we introduce a new longest possible secondary structure, sequence repeat structure, base missing structure, and spatial distance feature based on the existing sequence pairing feature, Manhattan distance feature, and RNA coding matrix, and fully exploit the structural and distance features and propose a more comprehensive structural vision feature; and (2) we pre-train the structural vision feature and combine it with the sequence feature to construct a multimodal model. The self-attention module and the cross-attention module fully integrate two-modal features for various downstream analysis.
At present, few studies have combined the vision feature with RNA structural information and the sequence feature to build the multimodal model. Our DRFormer model integrates RNA sequence module and structural vision module in a multimodal way, bringing excellent performance and generalization potential to downstream RNA tasks. For example, it outperforms existing advanced technologies in RNA downstream tasks, such as RNA secondary structure prediction, protein–RNA interaction prediction, and RNA classification. At the same time, it also achieves satisfactory results in generalized DNA downstream experiments.
Although the proposed DRFormer has achieved the satisfactory performance on various RNA downstream analyses, there are still some problems to be improved in future. For example, in RNA downstream tasks such as the sequence classification, protein–RNA interaction prediction, and RNA secondary structure prediction, the entire sequence can be input into our model and the prediction is slightly better than our current result. But the computational complexity of our model will significantly increase. Therefore, the sequence length is limited to 112, considering the great equipment and time requirement. How to combine the overlapping sequence windows and hierarchical strategies with our current model to improve the prediction performance will be our future work. In addition, when using the SWIN-RNA pre-trained model for calculation, this model can expand the scaling range through the Patch Embed (PE) layer, which can speed up the model training, but this would cause some kind of accuracy impact. At the same time, the structural features that we designed are implemented based on the standard pairing method [
22], which may miss some pseudo-structures (the cross-pairing base pair information in the pseudoknot or the contribution of atypical pairing regions), thereby affecting the acquisition of structure and the performance of the model. In addition, we find that the longest possible secondary structure, sequence repeat structure, base missing structure, and spatial distance feature have been verified to be effective in RNA sequence analysis. How to expand these new structural and distance features and apply them to other sequence analyses will also be our future work.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}