
Metabolomic and Transcriptomic Analyses of Flavonoid Biosynthesis in Dendrobium devonianum Flowers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Metabolite Extraction
2.3. HPLC-MS/MS Analysis
2.4. Metabolites Identification and Quantification
2.5. RNA Sequencing
2.6. Analysis of RNA Sequencing Data
2.7. Quantitative Real-Time PCR
3. Results
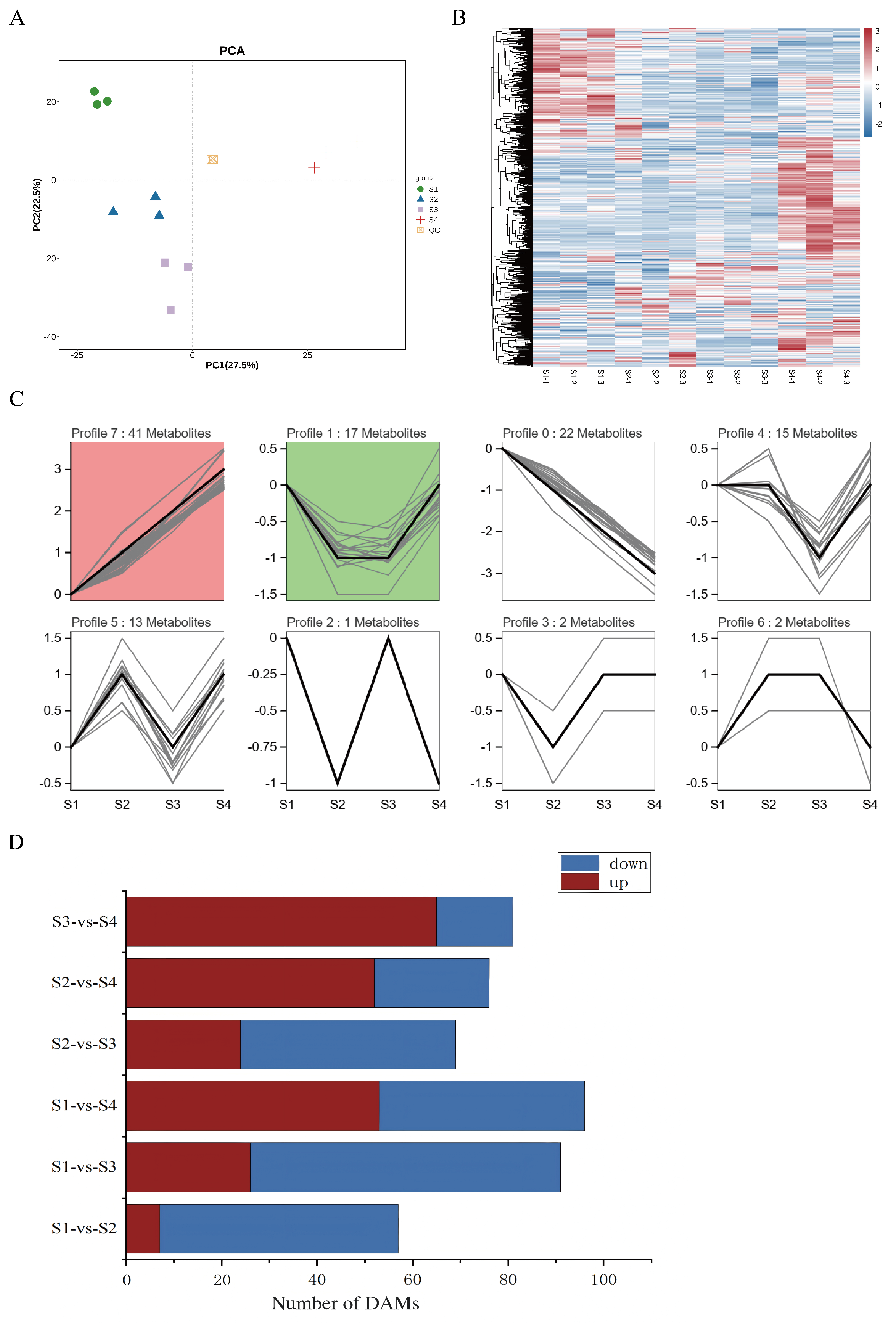
3.1. Metabolite Profiling of Four Developmental Periods of D. devonianum Flowers
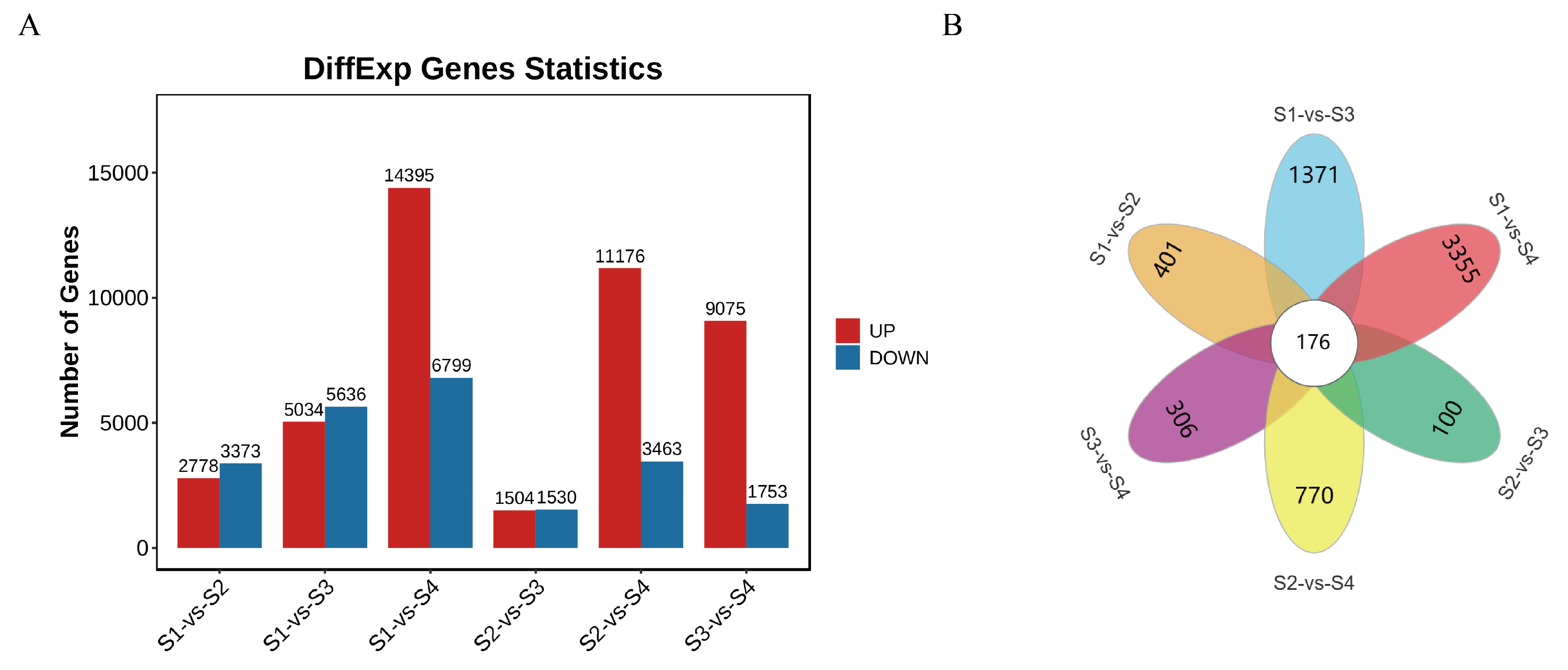
3.2. Transcriptomic Analysis in Four Developmental Stages of D. devonianum Flowers
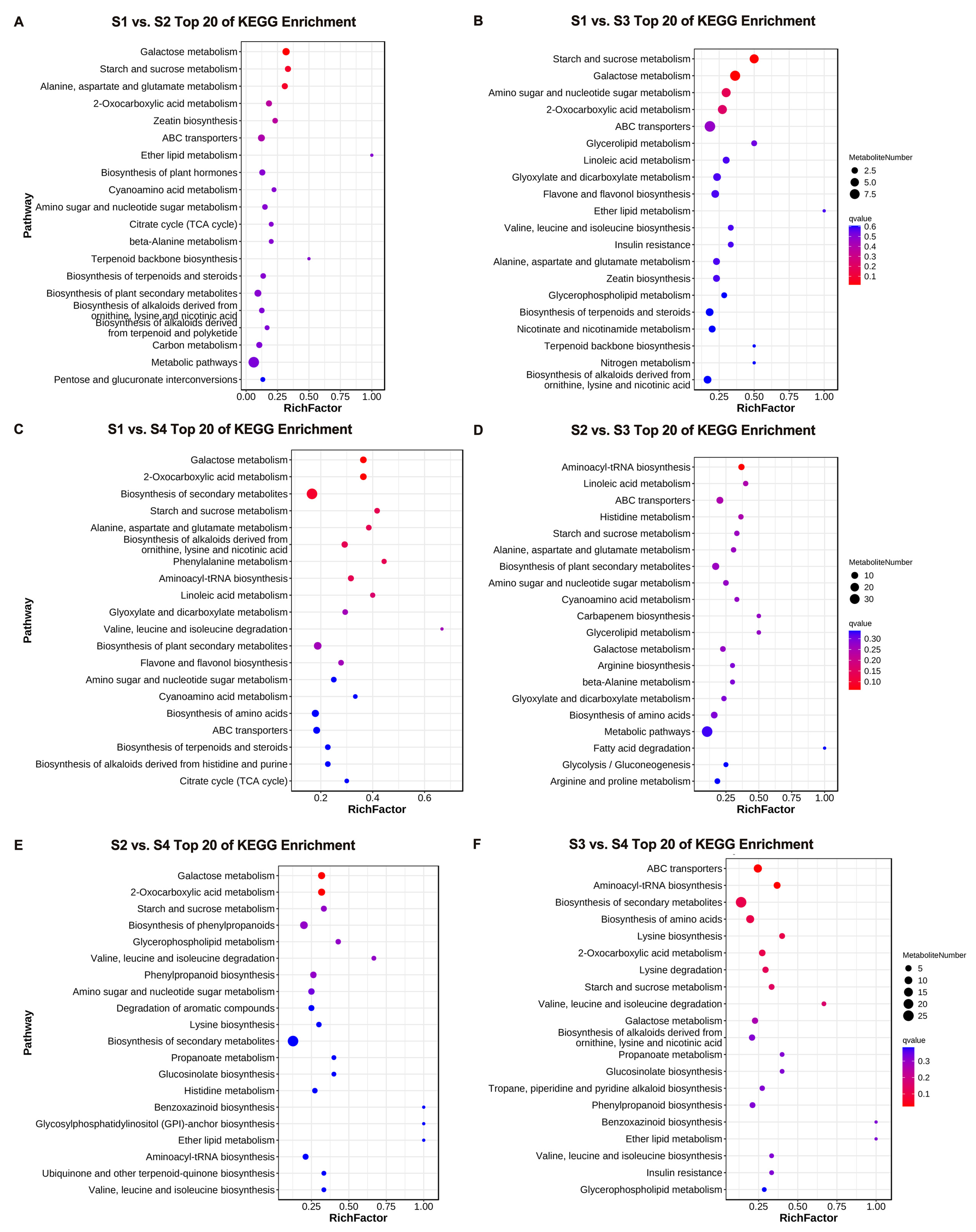
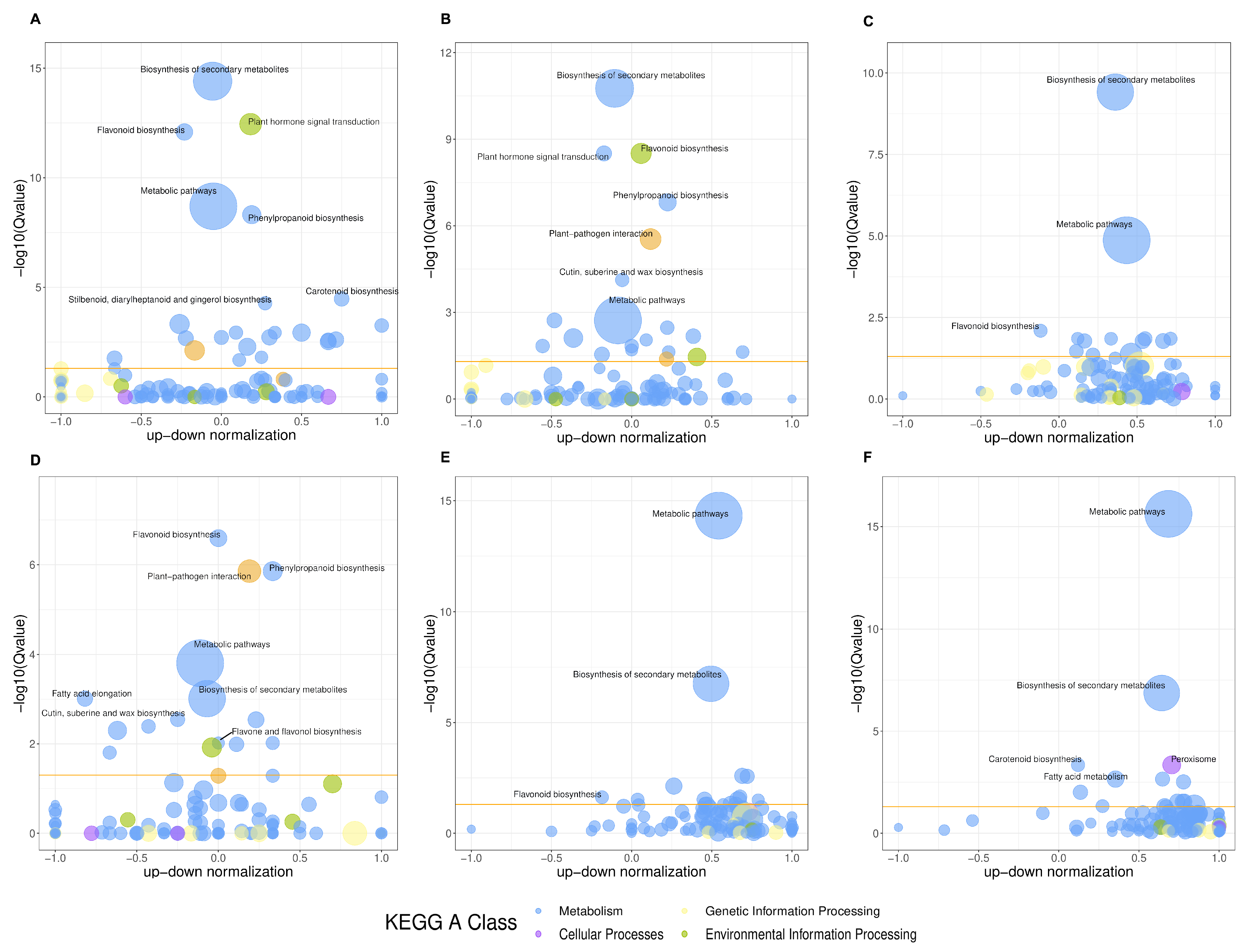
3.3. GO Enrichment and KEGG Pathway Analysis of DEGs
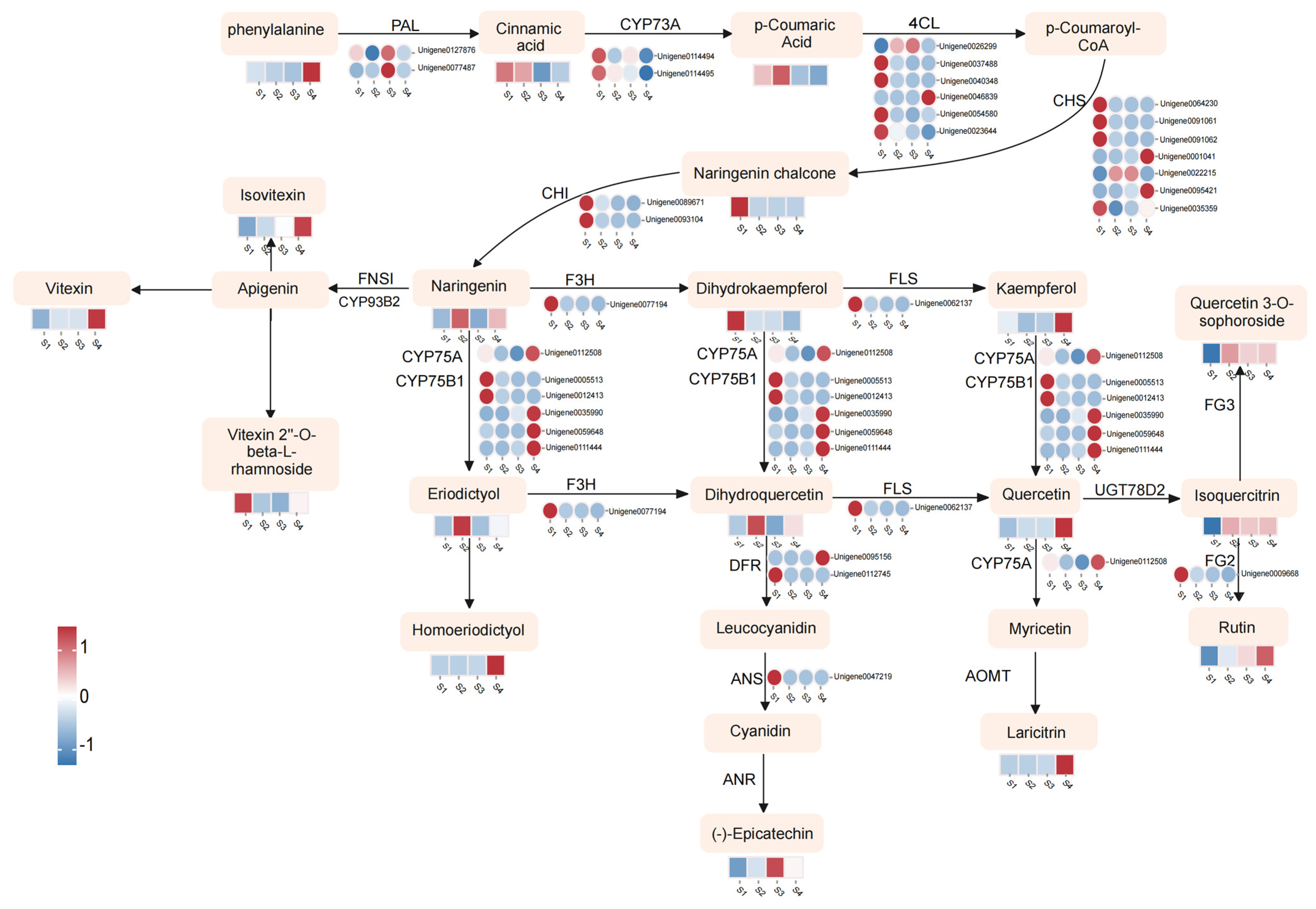
3.4. Metabolic Changes and Transcriptome Regulation Associated with Flavonoid Biosynthesis
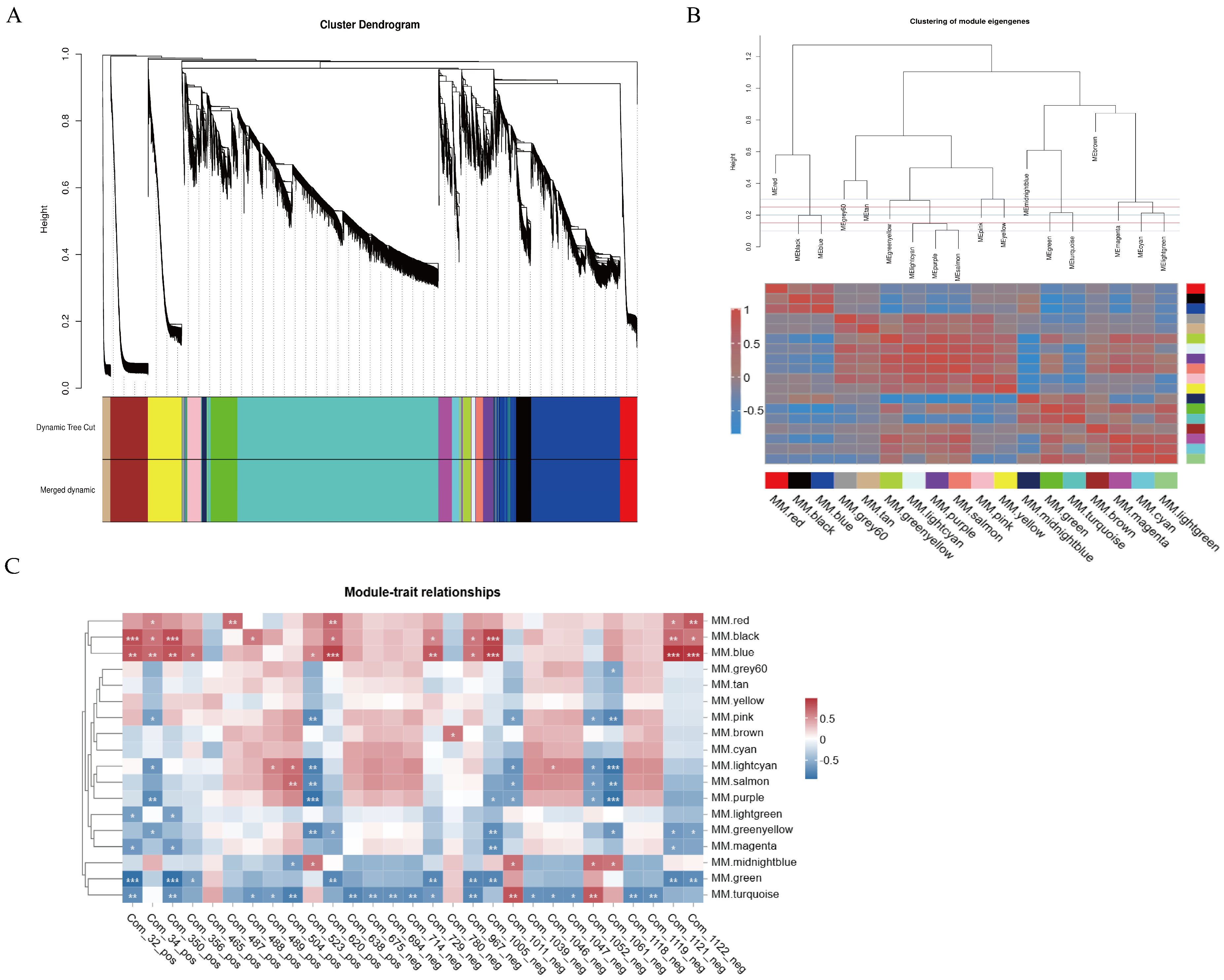
3.5. Gene Co-Expression Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Zhao, T.; Liu, J.; Zhao, R.; Zheng, S.; Chun, Z.; Hu, Y. Advances in chemical compounds and pharmacological effects of Dendrobii Caulis. Chin. Tradit. Herb. Drugs 2018, 49, 9. [Google Scholar]
- Wang, Y.H. Traditional Uses and Pharmacologically Active Constituents of Dendrobium Plants for Dermatological Disorders: A Review. Nat. Prod. Bioprospect. 2021, 11, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Dong, Q.; Zhou, L.; Zhou, L.; Tan, W. Investigation of Main Medicinal Dendrobium Resource in Yunnan. Yunnan J. Tradit. Chin. Med. Mater. Med. 2016, 37, 3. [Google Scholar]
- Song, B. Exploration on the introduction and domestication cultivation technology of wild Dendrobium devonianum in Yunnan Province. J. Smart Agric. 2022, 2, 72–74. [Google Scholar]
- Wang, H.; Li, M.; Hao, J.; Cheng, L.; Ma, G. Research Advances of Dendrobium devonianum. Chin. Arch. Tradit. Chin. Med. 2014, 32, 2732–2735. [Google Scholar]
- Wu, L.; Ding, Y.; Xue, Y.; Cheng, D.; Xu, H. Research on bibenzyl constituents and TLC identification of the stem of Dendrobium devoninum. Northwest Pharm. J. 2020, 35, 791–795. [Google Scholar]
- Wu, L.; Lu, Y.; Ding, Y.; Zhao, J.; Xu, H.; Chou, G. Four new compounds from Dendrobium devonianum. Nat. Prod. Res. 2019, 33, 2160–2168. [Google Scholar] [CrossRef]
- Lai, Y. Study on the efficacy of Dendrobium flowers. Technol. Trend 2020, 18, 277. [Google Scholar]
- Dong, S.; Ren, Y.; Zhang, X.; Wang, H.; Lu, J.; Hu, Y. Optimization of Extraction Process and Antioxidant Activity of Total Flavonoids in Dendrobium devoninum Flower. Farm. Prod. Process. 2023, 9, 27–32+36. [Google Scholar]
- Qu, J.; He, Y.; Sun, Z.; Su, J.; Liao, X.; Yang, J. Comparison of Amino Acids and Volatile Constituents in Four Kinds of Dendrobium Flowers. Mod. Chin. Med. 2018, 20, 387–394. [Google Scholar]
- Luo, H.; Liu, Q.S.; Fan, J.; Li, C.; Zhao, J.; Zhao, M. Determination of Nutrients in Flower of Dendrobium devonianum. Chin. Wild Plant Resour. 2021, 40, 18–22. [Google Scholar]
- Wang, D.; Fan, B.; Wang, Y.; Zhang, L.; Wang, F. Optimum Extraction, Characterization, and Antioxidant Activities of Polysaccharides from Flowers of Dendrobium devonianum. Int. J. Anal. Chem. 2018, 2018, 3013497. [Google Scholar] [CrossRef]
- Li, C.; Liu, Q.; Liu, K.; Liang, Z.; Liu, M.; Zhang, Z.; Cheng, J.; Zhao, J.; An, J.; Li, H. Chemical Compounds and Antioxidant Activities in Four Kinds of Dendrobium’s Flower. Sci. Technol. Eng. 2021. [Google Scholar]
- Chan, C.-F.; Wu, C.-T.; Huang, W.-Y.; Lin, W.-S.; Wu, H.-W.; Huang, T.-K.; Chang, M.-Y.; Lin, Y.-S. Antioxidation and Melanogenesis Inhibition of Various Dendrobium tosaense Extracts. Molecules 2018, 23, 1810. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Liu, J.; Liang, J.; Si, J.; Wu, S. Dendrobium officinale leaves as a new antioxidant source. J. Funct. Foods 2017, 37, 400–415. [Google Scholar] [CrossRef]
- Liao, X.; Xie, Z.; Liang, Z.; Zhou, Z.; Huang, Y.; Wei, G. Determination of five flavonoid glycosides in Dendrobium dentatum. J. Chin. Med. Mater. 2018, 41, 2863–2865. [Google Scholar]
- Shen, Y.; Zhou, Z.; Yang, Y.; Zhao, X.; Li, A. Chemical Constituents from Stem of Dendrobium devonianum. Nat. Prod. Res. Dev. 2012, 24, 339–341. [Google Scholar]
- Zhao, M.; Fan, J.; Liu, Q.; Luo, H.; Tang, Q.; Li, C.; Zhao, J.; Zhang, X. Phytochemical profiles of edible flowers of medicinal plants of Dendrobium officinale and Dendrobium devonianum. Food Sci. Nutr. 2021, 9, 6575–6586. [Google Scholar] [CrossRef]
- Qi, K.; Wu, X.; Gao, X.; Li, L.; Xie, Z.; Gu, C.; Zhang, S. Metabolome and transcriptome analyses unravel the inhibition of embryo germination by abscisic acid in pear. Sci. Hortic. 2022, 292, 110652. [Google Scholar]
- David, A.V.A.; Arulmoli, R.; Parasuraman, S. Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacogn. Rev. 2016, 10, 84–89. [Google Scholar]
- Wang, Z.; Wan, H.; Tong, X.; He, Y.; Yang, J.; Zhang, L.; Shao, C.; Ding, Z.; Wan, H.; Li, C. An integrative strategy for discovery of functional compound combination from Traditional Chinese Medicine: Danhong Injection as a model—ScienceDirect. Biomed. Pharmacother. 2021, 138, 111451. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liao, Y.; Teixeira da Silva, J.A.; Yang, Z.; Duan, J. Differential Accumulation of Anthocyanins in Dendrobium officinale Stems with Red and Green Peels. Int. J. Mol. Sci. 2018, 19, 2857. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, Z.; Song, Z.; Shi, X.; Liu, C.; Chen, Q.; Liu, H. Integrated transcriptomic and metabolomic analysis reveal the molecular and metabolic basis of flavonoids in Chinese sour jujube fruits in different harvest times. Postharvest Biol. Technol. 2024, 209, 112685. [Google Scholar] [CrossRef]
- Chen, J.; Tang, W.; Li, C.; Kuang, D.; Xu, X.; Gong, Y.; Liu, F.; Gao, S. Multi-omics analysis reveals the molecular basis of flavonoid accumulation in fructus of Gardenia (Gardenia jasminoides Ellis). BMC Genom. 2023, 24, 588. [Google Scholar] [CrossRef]
- Shen, C.; Guo, H.; Chen, H.; Shi, Y.; Meng, Y.; Lu, J.; Feng, S.; Wang, H. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci. Rep. 2017, 7, 187. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, S.; Yu, Z.; Zheng, X.; Liang, S.; Ren, H.; Qi, X. Transcription-Associated Metabolomic Analysis Reveals the Mechanism of Fruit Ripening during the Development of Chinese Bayberry. Int. J. Mol. Sci. 2024, 25, 8654. [Google Scholar] [CrossRef]
- Wang, W.; Yu, H.; Li, T.; Li, L.; Zhang, G.; Liu, Z.; Huang, T.; Zhang, Y. Comparative Proteomics Analyses of Pollination Response in Endangered Orchid Species Dendrobium Chrysanthum. Int. J. Mol. Sci. 2017, 18, 2496. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Biogeosciences 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Zhang, Z.; Luo, C.; Wei, K.; Li, X.; Mu, X.; Duan, M.; Zhu, C.; Jin, L.; He, X. MultiPrime: A reliable and efficient tool for targeted next-generation sequencing. iMeta 2023, 2, e143. [Google Scholar] [CrossRef]
- Ghasemzadeh, A.; Ghasemzadeh, N. Flavonoids and phenolic acids: Role and biochemical activity in plants and human. J. Med. Plants Res. 2011, 2011, 6697–6703. [Google Scholar] [CrossRef]
- Lobstein, A.; Rietsch-Jako, L.; Haag-Berrurier, M.; Anton, R. Seasonal Variations of the Flavonoid Content from Ginkgo biloba Leaves. Planta Med. 1991, 57, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, X.; Peng, X.; Wang, W.; Zhao, C.; Zu, Y.; Fu, Y. Fast and green extraction and separation of main bioactive flavonoids from Radix Scutellariae. Ind. Crops Prod. 2015, 63, 175–181. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chang, E.J.; Kim, H.J.; Park, J.H.; Choi, S.W. Antioxidative flavonoids from leaves of Carthamus tinctorius. Arch. Pharmacal Res. 2002, 25, 313–319. [Google Scholar] [CrossRef]
- Su, L.; Wu, M.; Zhang, T.; Zhong, Y.; Cheng, Z.M. Identification of key genes regulating the synthesis of quercetin derivatives in Rosa roxburghii through integrated transcriptomics and metabolomics. J. Integr. Agric. 2024, 23, 876–887. [Google Scholar] [CrossRef]
- Borredá, C.; Perez-Roman, E.; Talon, M.; Terol, J. Comparative transcriptomics of wild and commercial Citrus during early ripening reveals how domestication shaped fruit gene expression. BMC Plant Biol. 2022, 22, 123. [Google Scholar] [CrossRef]
- Routaboul, J.-M.; Kerhoas, L.; Debeaujon, I.; Pourcel, L.; Caboche, M.; Einhorn, J.; Lepiniec, L. Flavonoid diversity and biosynthesis in seed of Arabidopsis thaliana. Planta 2006, 224, 96–107. [Google Scholar] [CrossRef]
- Yuan, Y.; Zuo, J.; Zhang, H.; Zu, M.; Yu, M.; Liu, S. Transcriptome and metabolome profiling unveil the accumulation of flavonoids in Dendrobium officinale. Genomics 2022, 114, 110324. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.; Šamec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnol. Adv. 2020, 38, 107316. [Google Scholar] [CrossRef] [PubMed]
- Vianello, A.; Patui, S.; Bertolini, A.; Peresson, C.; Braidot, E.; Zancani, M.; Petrussa, E. Plant Flavonoids—Biosynthesis, Transport and Involvement in Stress Responses. Int. J. Mol. Sci. 2013, 14, 14950–14973. [Google Scholar] [CrossRef]
- Shi, M.Z.; Xie, D.Y. Biosynthesis and metabolic engineering of anthocyanins in Arabidopsis thaliana. Recent Pat. Biotechnol. 2014, 8, 47–60. [Google Scholar] [CrossRef]
- Cheng, A.X.; Han, X.J.; Wu, Y.F.; Lou, H.X. The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis. Int. J. Mol. Sci. 2014, 15, 1080–1095. [Google Scholar] [CrossRef]
- Gao, J.; Ren, R.; Wei, Y.; Jin, J.; Zhu, G. Comparative Metabolomic Analysis Reveals Distinct Flavonoid Biosynthesis Regulation for Leaf Color Development of Cymbidium sinense ‘Red Sun’. Int. J. Mol. Sci. 2020, 21, 1869. [Google Scholar] [CrossRef]
- Jiang, T.; Guo, K.; Liu, L.; Tian, W.; Wen, C. Integrated transcriptomic and metabolomic data reveal the flavonoid biosynthesis metabolic pathway in Perilla frutescens (L.) leaves. Sci. Rep. 2020, 10, 16207. [Google Scholar] [CrossRef]
- Raso, G.M.; Meli, R.; Carlo, G.D.; Pacilio, M.; Carlo, R.D. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci. 2001, 68, 921–931. [Google Scholar] [CrossRef]
- Sun, W.; Meng, X.; Liang, L.; Jiang, W.; Huang, Y.; He, J.; Hu, H.; Almqvist, J.; Gao, X.; Wang, L. Molecular and Biochemical Analysis of Chalcone Synthase from Freesia hybrid in Flavonoid Biosynthetic Pathway. PLoS ONE 2015, 10, e0119054. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Chaudhary, P.R.; Bang, H.; Jayaprakasha, G.K.; Patil, B.S. Variation in key flavonoid biosynthetic enzymes and phytochemicals in Rio Red grapefruit (Citrus paradisi Macf) during fruit development. J. Agric. Food Chem. 2016, 64, 9022–9032. [Google Scholar] [CrossRef] [PubMed]
- Britsch, L.; Britsch, L.; Grisebach, H. Purification and characterization of (2S)-flavanone 3-hydroxylase from Petunia hybrida. Eur. J. Biochem. 1986, 156, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Yamada, E.; Saito, M.; Fujita, K.; Takahashi, H.; Nakatsuka, T. Molecular characterization of mutations in white-flowered torenia plants. BMC Plant Biol. 2014, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wu, Z.; Yang, G.; Lu, S.; Jia, Y.; Wu, Y.; Zhou, R.; Xie, X. Cloning and functional analysis of flavanone 3-hydroxylase gene in Rhododendron hybridum Hort. Chin. J. Biotechnol. 2023, 39, 653–669. [Google Scholar]
- Forkmann, G.; Stotz, G. Selection and characterisation of flavanone 3-hydroxylase mutants of Dahlia, Streptocarpus, Verbena and Zinnia. Planta 1984, 161, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Sun, P. The Study of Arabidopsis Flavanone-3-hydroxylase in Regulating Abiotic Stress Tolerance. Bachelor’s Thesis, Shandong Agricultural University, Shandong, China, 2014. [Google Scholar]
- Yuan, Y.; Zuo, J.; Zhang, H.; Zu, M.; Liu, S. Analysis of the different growth years accumulation of flavonoids in Dendrobium moniliforme (L.) Sw. by the integration of metabolomic and transcriptomic approaches. Front. Nutr. 2022, 9, 928074. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, S.; Fu, H.; Zhou, C.; Chen, L.; Li, X.; Lin, Y.; Lai, Z.; Guo, Y. Transcriptome and Phytochemical Analyses Provide New Insights Into Long Non-Coding RNAs Modulating Characteristic Secondary Metabolites of Oolong Tea (Camellia sinensis) in Solar-Withering. Front. Plant Sci. 2019, 10, 1638. [Google Scholar] [CrossRef]
- Tanaka, Y. Flower colour and cytochromes P450. Phytochem. Rev. 2006, 5, 283–291. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wu, Y.; Pu, R.; Li, X.; Bai, T.; Li, N.; Zhou, Y.; Zhang, J. Metabolomic and Transcriptomic Analyses of Flavonoid Biosynthesis in Dendrobium devonianum Flowers. Genes 2025, 16, 264. https://doi.org/10.3390/genes16030264
Li Y, Wu Y, Pu R, Li X, Bai T, Li N, Zhou Y, Zhang J. Metabolomic and Transcriptomic Analyses of Flavonoid Biosynthesis in Dendrobium devonianum Flowers. Genes. 2025; 16(3):264. https://doi.org/10.3390/genes16030264
Chicago/Turabian StyleLi, Yue, Yawen Wu, Ran Pu, Xuejiao Li, Tian Bai, Nengbo Li, Ying Zhou, and Jingli Zhang. 2025. "Metabolomic and Transcriptomic Analyses of Flavonoid Biosynthesis in Dendrobium devonianum Flowers" Genes 16, no. 3: 264. https://doi.org/10.3390/genes16030264
APA StyleLi, Y., Wu, Y., Pu, R., Li, X., Bai, T., Li, N., Zhou, Y., & Zhang, J. (2025). Metabolomic and Transcriptomic Analyses of Flavonoid Biosynthesis in Dendrobium devonianum Flowers. Genes, 16(3), 264. https://doi.org/10.3390/genes16030264