
Genome Sequencing Reveals the Potential of Enterobacter sp. Strain UNJFSC003 for Hydrocarbon Bioremediation

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Strain, Extraction of DNAg, and Genome Sequencing
2.2. Genome Assembly, Annotation, and Functional Analysis
2.3. Comparative Genome Analysis, Molecular and Pangenome Confirmation
2.4. Prediction of Hydrocarbon-Degrading Genes and Enzymes
2.5. In Silico Protein–Protein Interaction and Subcellular Localization of Hydrocarbon-Degrading Proteins
2.6. Molecular Modeling, Model Validation, and Molecular Docking
3. Results
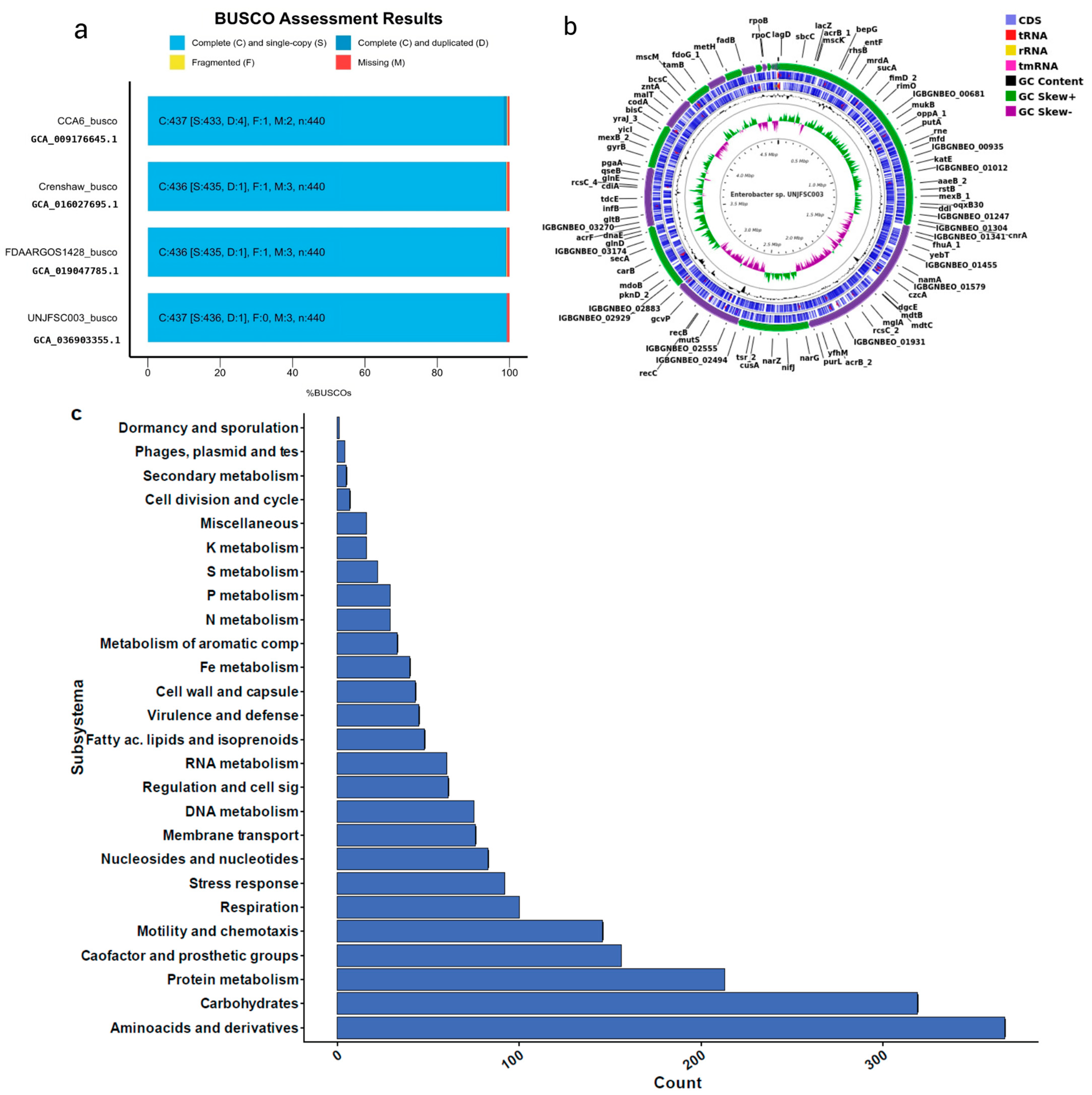
3.1. Isolation of the Strain and Genome Characteristics of Enterobacter sp. UNJFSC 003
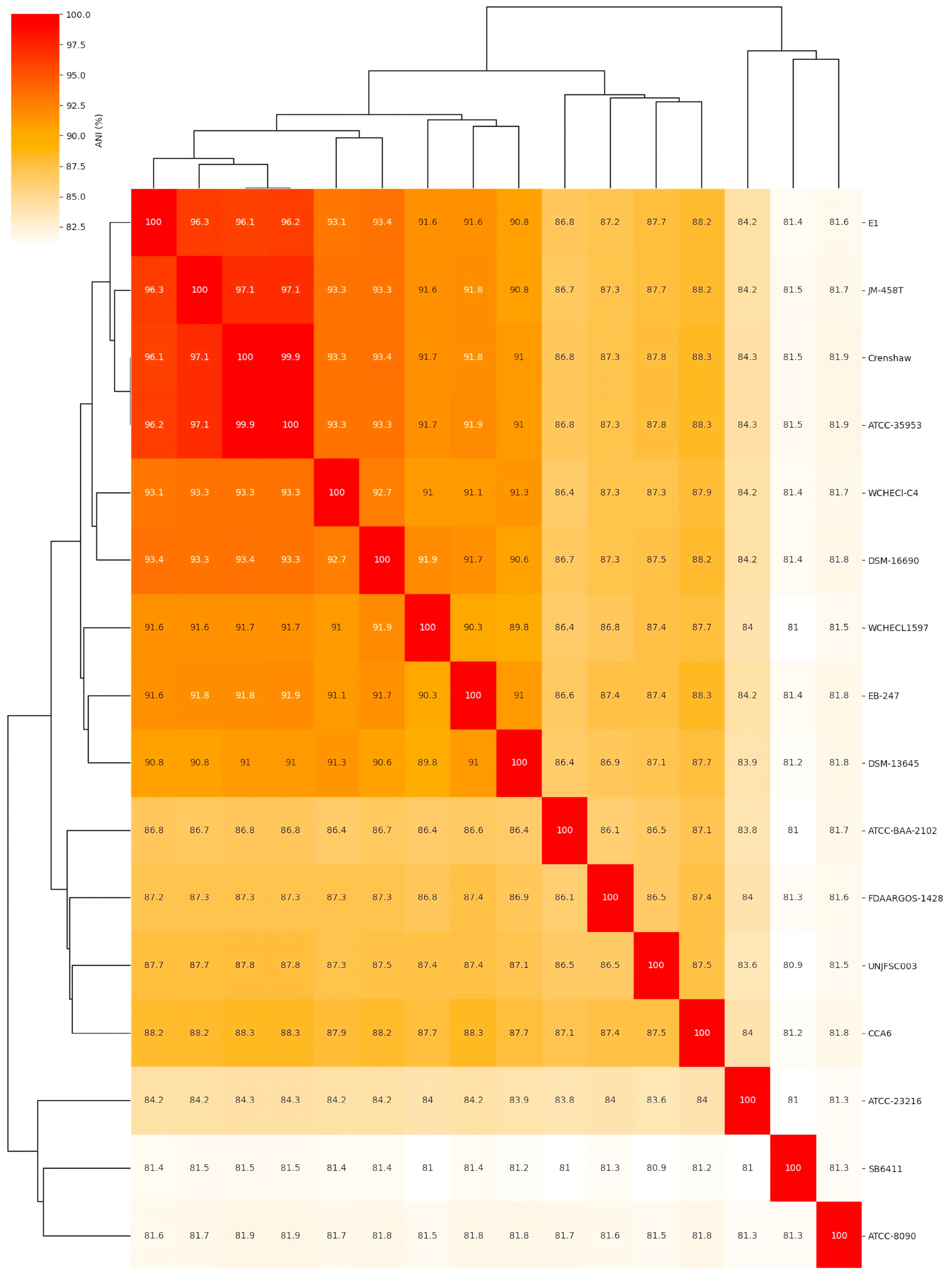
3.2. Comparative Analysis of the Complete Genome of Enterobacter sp. UNJFSC 003
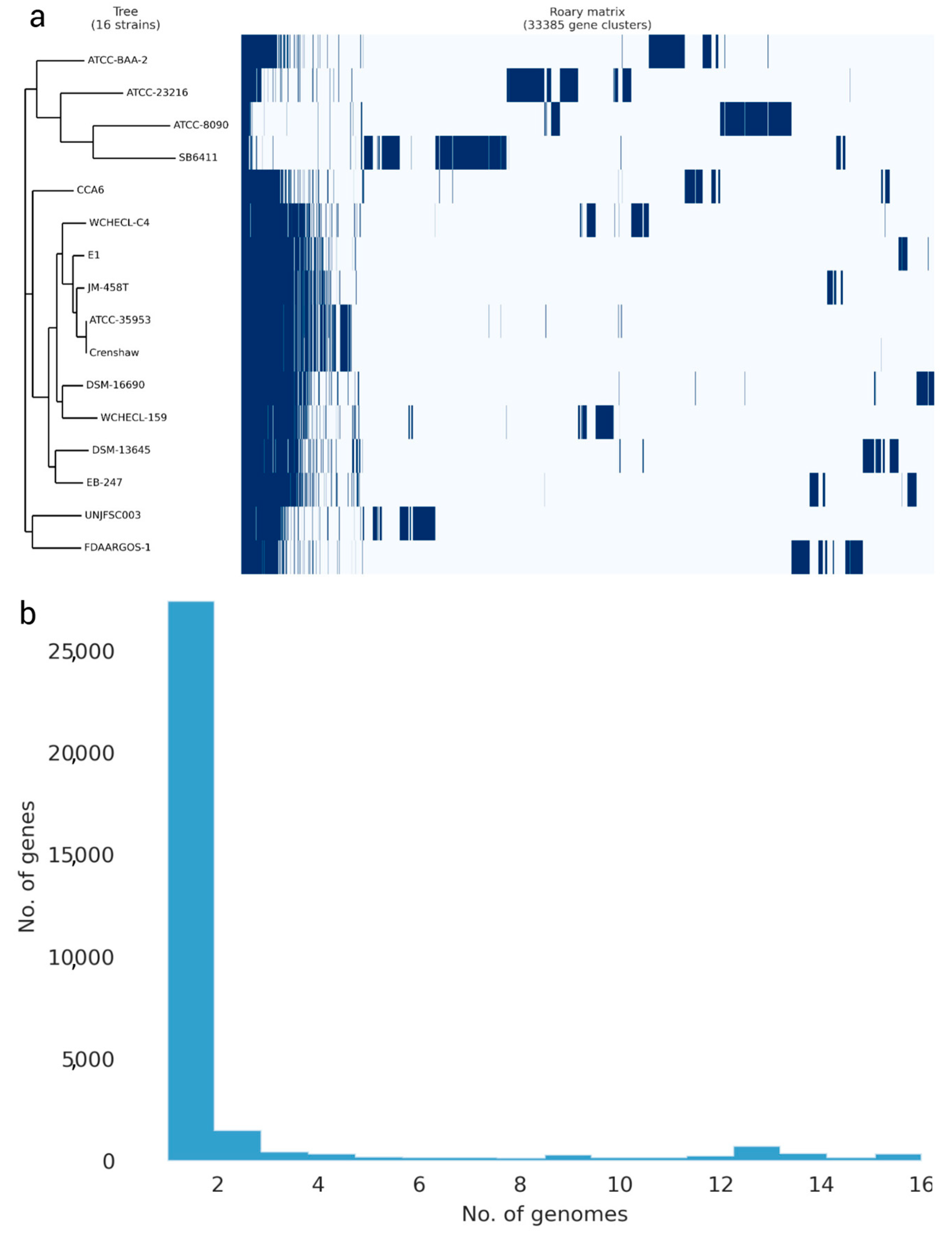
3.3. Analysis of the Pangenome of Enterobacter sp. UNJFSC 003
3.4. The Strain UNJFSC 003 Harbors Genes Encoding Enzymes for the Bioremediation of Hydrocarbons
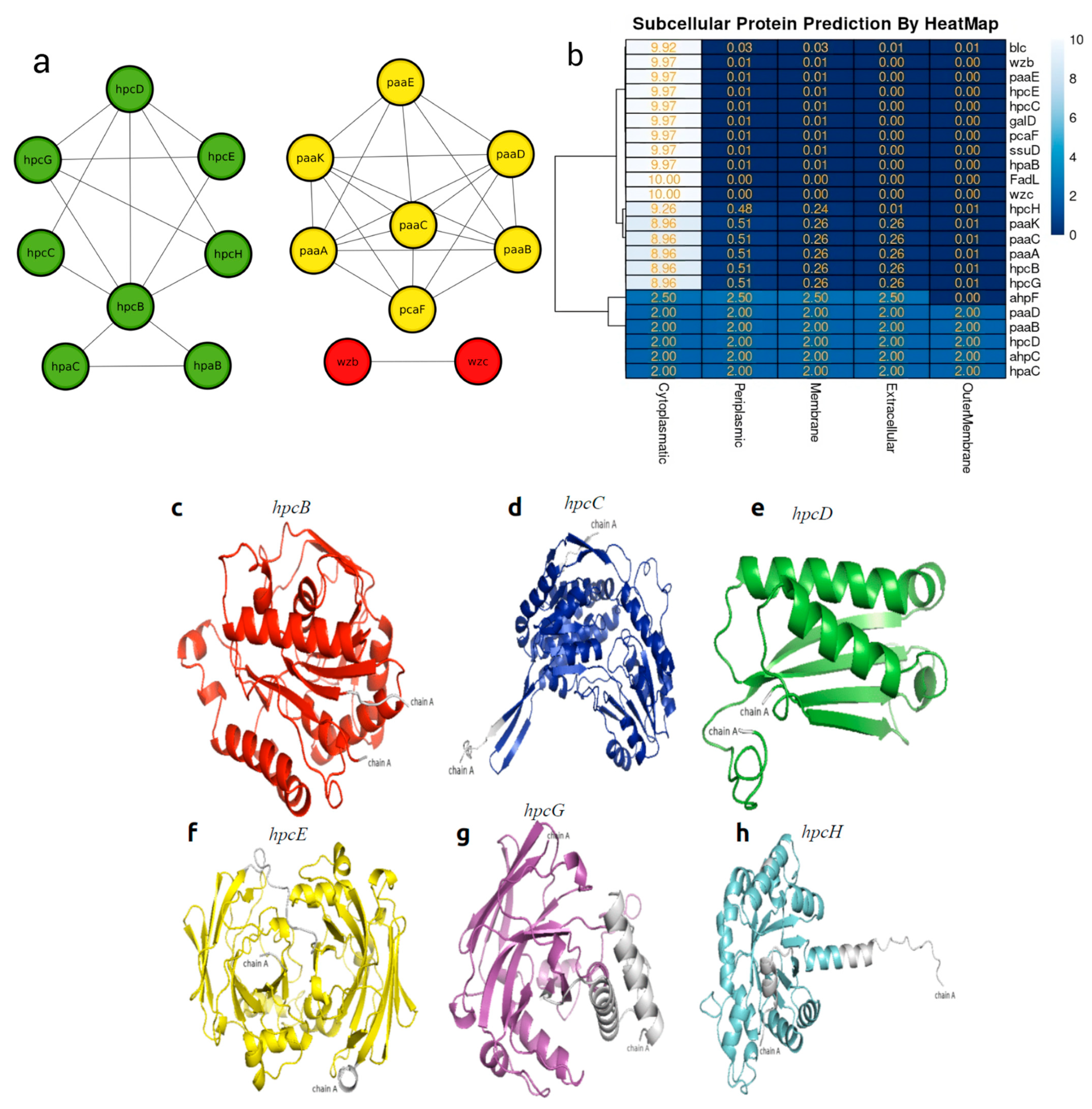
3.5. In Silico Protein–Protein Interaction and Heat Map of Proteins Involved in Bioremediation
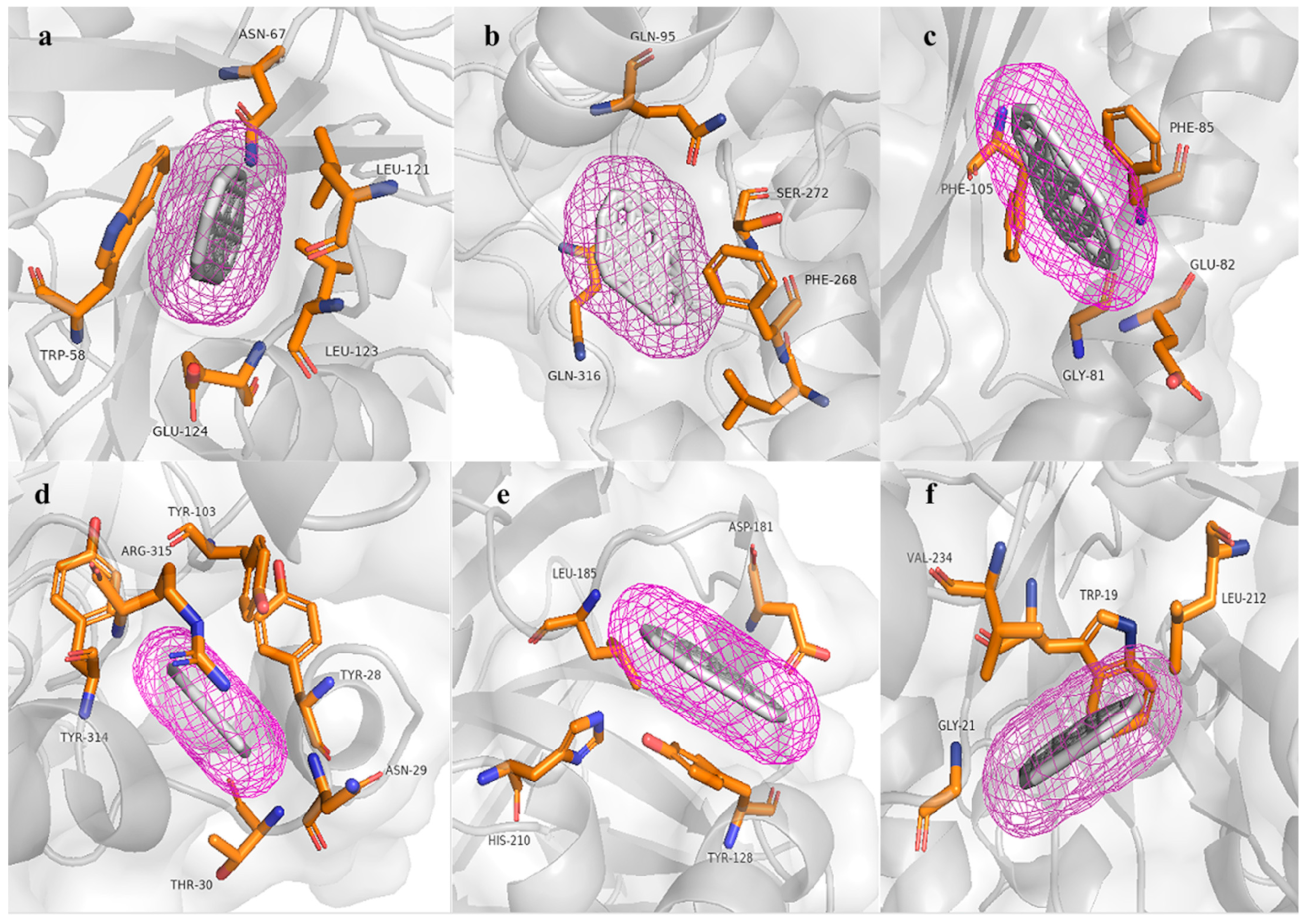
3.6. Molecular Modeling and Docking Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, K.; Chandra, S. Treatment of petroleum hydrocarbon polluted environment through bioremediation: A review. Pak. J. Biol. Sci. 2013, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tormoehlen, L.M.; Tekulve, K.J.; Ñañagas, K.A. Hydrocarbon toxicity: A review. Clin. Toxiclogy 2014, 52, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Volkman, J.K. Hydrocarbons. In Geochemistry. Encyclopedia of Earth Science; Springer: Dordrecht, The Netherlands, 1998. [Google Scholar] [CrossRef]
- Das, T.; Dash, H.R. Microbial Bioremediation: A Potential Tool for Restoraton of Contaminated Areas. In Microbial Biodegradation and Biorremediation; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–21. [Google Scholar] [CrossRef]
- Ayilara, M.S.; Babalola, O.O. Bioremediation of environmental wastes: The role of microorganisms. Front. Agron. 2023, 5, 1–15. [Google Scholar] [CrossRef]
- Castillo-Rogel, R.T.; More-Calero, F.J.; La-Torre, M.C.; Fernandez-Ponce, J.N.; Mialhe-Matonnier, E.L. Aislamiento de bacterias con potencial biorremediador y analisis de comunidades bacterianas de zona impactada por derrame de petroleo encondorcanqui-Amazonas-Peru. Rev. Investig. Altoand. 2020, 22, 215–225. [Google Scholar] [CrossRef]
- Emenike, C.U.; Agamuthu, P.; Fauziah, S.H.; Omo-Okoro, P.N.; Jayanthi, B. Enhanced Bioremediation of Metal-Contaminated Soil by Consortia of Proteobacteria. Water Air Soil Pollut. 2023, 234, 731. [Google Scholar] [CrossRef]
- Nyoyoko, V.F. Chapter 13—Proteobacteria response to heavy metal pollution stress and their bioremediation potential. In Cost Effective Technologies for Solid Waste and Wastewater Treatment; Elsevier: Amsterdam, The Netherlands, 2022; pp. 147–159. [Google Scholar] [CrossRef]
- Thacharodi, A.; Hassan, S.; Singh, T.; Mandal, R.; Chinnadurai, J.; Khan, H.A.; Hussain, M.A.; Brindhadevi, K.; Pugazhendhi, A. Biormediation of polycyclic aromatic hydrocarbons: Anupdated microbiological review. Chemosphere 2023, 328, 138498. [Google Scholar] [CrossRef]
- Kugarajah, V.; Nisha, K.N.; Jayakumar, R.; Sahabudeen, S.; Ramakrishnan, P.; Mohamed, S.B. Significance of microbial genome in environmental remediation. Microbiol. Res. 2023, 271, 127360. [Google Scholar] [CrossRef]
- Perruchon, C.; Chatzinotas, A.; Omirou, M.; Vasileiadis, S.; Menkissoglou-Spiroudi, U.; Karpouzas, D.G. Isolation of a bacterial consortium able to degrade the fungicide thiabendazole: The key role of a Sphingomonas phylotype. Appl. Microbiol. Biotechnol. 2017, 101, 3881–3893. [Google Scholar] [CrossRef]
- Villela, H.; Modolon, F.; Schultz, J.; Delgadillo-Ordoñez, N.; Carvalho, S.; Soriano, A.U.; Peixoto, R.S. Genome analysis of a coral-associated bacterial consortium highlights complementary hydrocarbon degradation ability and other beneficial mechanisms for the host. Sci. Rep. 2023, 13, 12273. [Google Scholar] [CrossRef]
- Joutey, N.T.; Bahafid, W.; Sayel, H.; El Ghachtouli, N. Biodegradation: Involved Microorganisms and Genetically Engineered Microorganisms. In Biodegradation—Life of Science; Chamy, R., Ed.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, S.P.; Iqbal, H.M.; Tong, Y.W. Omics approaches in bioremediation of environmental contaminants: An integrated approach for environmental safety and sustainability. Environ. Res. 2022, 211, 113102. [Google Scholar] [CrossRef]
- Mishra, M.; Singh, S.K.; Kumar, A. Chapter 32—Role of omics approaches in microbial bioremediation. In Microbe Mediated Remediation of Environmental Contaminants; Kumar, A., Singh, V.K., Singh, P., Mishra, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 435–445. [Google Scholar] [CrossRef]
- Wu, J.; Lv, J.; Zhao, L.; Zhao, R.; Gao, T.; Xu, Q.; Liu, D.; Yu, Q.; Ma, F. Exploring the role of microbial proteins in controlling environmental pollutants based on molecular simulation. Sci. Total. Environ. 2023, 905, 167028. [Google Scholar] [CrossRef] [PubMed]
- Alav, I.; Kobylka, J.; Kuth, M.S.; Pos, K.M.; Picard, M.; Blair, J.M.A.; Bavro, V.N. Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria. Chem. Rev. 2021, 121, 5479–5596. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Maheshwari, D.K. Insights into zinc-sensing metalloregulator ‘Zur’ deciphering mechanism of zinc transportation in Bacillus spp. by modeling, simulation and molecular docking. J. Biomol. Struct. Dyn. 2020, 40, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Kumari, P.; Kumar, Y. Chapter 19—Bioinformatics and computational tools in bioremediation and biodegradation of environmental pollutants. In Bioremediation for Environmental Sustainability; Kumar, V., Saxena, G., Shah, M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 421–444. [Google Scholar] [CrossRef]
- Morrison-Smith, S.; Boucher, C.; Sarcevic, A.; Noyes, N.; O’Brien, C.; Cuadros, N.; Ruiz, J. Challenges in large-scale bioinformatics projects. Humanit. Soc. Sci. Commun. 2022, 9, 125. [Google Scholar] [CrossRef]
- Arora, P.K.; Kumar, A.; Srivastava, A.; Garg, S.K.; Singh, V.P. Current bioinformatics tools for biodegradation of xenobiotic compounds. Front. Environ. Sci. 2022, 10, 980284. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 31 October 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008, 9, 75. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Manda, A.; Herman, E.K.; Chen, C.; Graham, M.; Domselaar, G.V.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Hunter, D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y. ANIclustermap: A Tool for Drawing ANI Clustermap Between All-vs-All Microbial Genomes. 2022. Available online: https://github.com/moshi4/ANIclustermap (accessed on 31 October 2024).
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden-Matth, T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Vargas, J.; Castelán-Sánchez, H.G.; Pardo-López, L. HADEG: A curated hydrocarbon aerobic degradation enzymes and genes database. Comput. Biol. Chem. 2023, 107, 107966. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeisz, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-) orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 31 October 2024).
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, W638–W646. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2018. Available online: https://github.com/raivokolde/pheatmap (accessed on 31 October 2024).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 31 October 2024).
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- de Beer, T.A.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Armenteros, J.J.A.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the Expasy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2024. Available online: http://www.pymol.org/pymol (accessed on 31 October 2024).
- Farmer, J.J.; Davis, B.R.; Hickman-Brenner, F.W.; McWhorter, A.; Huntley-Carter, G.P.; Asbury, M.A.; Riddle, C.; Wathen-Grady, H.G.; Elias, C.; Fanning, G.R. Biochemical identification of new species and biogroups of Enterobacteriaceae isolated from clinical specimens. J. Clin. Microbiol. 1985, 21, 46–76. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, 11. [Google Scholar] [CrossRef]
- Rosselló-Móra, R.; Amann, R. Past and future species definitions for bacteria and archaea. Syst. Appl. Microbiol. 2015, 38, 209–216. [Google Scholar] [CrossRef]
- Anzuay, M.S.; Prenollio, A.; Ludueña, L.M.; Morla, F.D.; Cerliani, C.; Lucero, C.; Angelini, J.G.; Taurian, T. Enterobacter sp. J49: A Native Plant Growth-Promoting Bacteria as Alternative to the Application of Chemical Fertilizers on Peanut and Maize Crops. Curr. Microbiol. 2023, 80, 85. [Google Scholar] [CrossRef]
- Tovar, C.; Sánchez Infantas, E.; Teixeira Roth, V. Plant community dynamics of lomas fog oasis of Central Peru after the extreme precipitation caused by the 1997-98 El Niño event. PLoS ONE 2018, 13, e0190572. [Google Scholar] [CrossRef]
- Muneeswari, R.; Iyappan, S.; Swathi, K.V.; Sudheesh, K.P.; Rajesh, T.; Sekaran, G.; Ramani, K. Genomic characterization of Enterobacter xiangfangensis STP-3: Application to real time petroleum oil sludge bioremediation. Microbiol. Res. 2021, 253, 126882. [Google Scholar] [CrossRef]
- Cao, Y.M.; Xu, L.; Jia, L.Y. Analysis of PCBs degradation abilities of biphenyl dioxygenase derived from Enterobacter sp. LY402 by molecular simulation. New Biotechnol. 2011, 29, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Hošková, M.; Ježdík, R.; Schreiberová, O.; Chudoba, J.; Šír, M.; Čejková, A.; Masák, J.; Jirků, V.; Řezanka, T. Structural and physiochemical characterization of rhamnolipids produced by Acinetobacter calcoaceticus, Enterobacter asburiae and Pseudomonas aeruginosa in single strain and mixed cultures. J. Biotechnol. 2015, 193, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, S.; Afzal, M.; Reichenauer, T.G.; Brady, C.L.; Sessitsch, A. Hydrocarbon degradation, plant colonization and gene expression of alkane degradation genes by endophytic Enterobacter ludwigii strains. Environ. Pollut. 2011, 159, 2675–2683. [Google Scholar] [CrossRef]
- Broderick, J.B. Catechol dioxygenases. Essays Biochem. 1999, 34, 173–189. [Google Scholar] [CrossRef]
- Baburam, C.; Feto, N.A. Mining of two novel aldehyde dehydrogenases (DHY-SC-VUT5 and DHY-G-VUT7) from metagenome of hydrocarbon contaminated soils. BMC Biotechnol. 2021, 21, 18. [Google Scholar] [CrossRef]
- Gangola, S.; Bhandari, G.; Joshi, S.; Sharma, A.; Simsek, H.; Bhatt, P. Esterase and ALDH dehydrogenase-based pesticide degradation by Bacillus brevis 1B from a contaminated environment. Environ. Res. 2023, 232, 116332. [Google Scholar] [CrossRef]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef]
- Roper, D.I.; Cooper, R.A. Purification, some properties and nucleotide sequence of 5-carboxymethyl-2-hydroxymuconate isomerase of Escherichia coli C. FEBS Lett. 1990, 266, 63–66. [Google Scholar] [CrossRef]
- Izumi, A.; Rea, D.; Adachi, T.; Unzai, S.; Park, S.-Y.; Roper, D.I.; Tame, J.R.H. Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. JMB 2007, 370, 899–911. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Feng, Y.; Zong, Z. Characterization of a strain representing a new Enterobacter species, Enterobacter chengduensis sp. nov. Antonie Leeuwenhoek 2018, 112, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.R.; Kondiah, K.; De Maayer, P. Complete Genome Sequence of Enterobacter xiangfangensis Pb204, a South African Strain Capable of Synthesizing Gold Nanoparticles. Microbiol. Resour. Announc. 2018, 7, e0086321. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.N.; Gazali, S.J.; Parthasarathy, A.; Wadsworth, C.B.; Miranda, R.R.; Thomas, B.N.; Hudson, A.O. Isolation, Whole-Genome Sequencing, and Annotation of Three Unclassified Antibiotic-Producing Bacteria, Enterobacter sp. Strain RIT 637, Pseudomonas sp. Strain RIT 778, and Deinococcus sp. Strain RIT 780. ASM J. Microbiol. Resour. Announc. 2021, 10, e00863-21. [Google Scholar] [CrossRef]
- Indugu, N.; Sharma, L.; Jackson, C.R.; Singh, P. Whole-Genome Sequence Analysis of Multidrug-Resistant Enterobacter hormaechei Isolated from Imported Retail Shrimp. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef]
- Szczerba, H.; Komon-Janczara, E.; Krawczyk, M.; Dudziak, K.; Nowak, A.; Kuzdraliński, A.; Waśko, A.; Targónski, Z. Genome analysis of a wild rumen bacterium Enterobacter aerogenes LU2—A novel bio-based succinic acid producer. Sci. Rep. 2020, 10, 1986. [Google Scholar] [CrossRef]
- Jahan, R.; Bodratti, A.M.; Tsianou, M.; Alexandridis, P. Biosurfactants, natural alternatives to synthetic surfactants: Physicochemical properties and applications. Adv. Colloid Interface Sci. 2020, 275, 102061. [Google Scholar] [CrossRef]
- Peng, X.; Zheng, Q.; Liu, L.; He, Y.; Li, T.; Jia, X. Efficient biodegradation of tetrabromobisphenol A by the novel strain Enterobacter sp. T2 with good environmental adaptation: Kinetics, pathways and genomic characteristics. J. Hazard. Mater. 2022, 429, 128335. [Google Scholar] [CrossRef]
- El-Beltagi, H.S.; Halema, A.; Almutairi, Z.M.; Almutairi, H.H.; Elarabi, N.; Abdelhadi, A.A.; Henawy, A.R.; Abdelhaleem, H.A.R. Draft genome analysis for Enterobacter kobei, a promising lead bioremediation bacterium. Front. Bioeng. Biotechnol. 2024, 11, 1335854. [Google Scholar] [CrossRef]
- Camacho, J.; Mesen-Porras, E.; Rojas-Gatjens, D.; Perez-Pantoja, D.; Puente-Sanchez, F.; Chavarria, M. Draft genome sequence of three hydrocarbon-degrading Pseudomonadota strains isolated from an abandoned century-old oil exploration well. ASM J. Environ. Microbiol. 2024, 13, e0107623. [Google Scholar] [CrossRef]
- Amin, A.; Naveed, M.; Sarwar, A.; Rasheed, S.; Saleem, H.G.M.; Latif, Z.; Bechthold, A. In vitro and in silico Studies Reveal Bacillus cereus AA-18 as a Potential Candidate for Bioremediation of Mercury-Contaminated Wastewater. Front. Microbiol. 2022, 13, 847806. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Gupta, A.; Singh, P.; Mishra, A.K.; Ranjan, R.K.; Srivastava, A. Siderophore-assisted cadmium hyperaccumulation in Bacillus subtilis. Int. Microbiol. 2019, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Laird, M.R.; Chen, F.; Rey, S.; Walsh, C.J.; Ester, M.; Brinkman, F.S.L. PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 2005, 21, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Peabody, M.A.; Lau, W.Y.V.; Hoad, G.R.; Jia, B.; Maguire, F.; Gray, K.L.; Beiko, R.G.; Brinkman, F.S.L. PSORTm: A bacteria and archaeal protein subcellular localization prediction tool for metagenomics data. Bioinformatics 2020, 36, 3043–3048. [Google Scholar] [CrossRef]
- Lou, F.; Okoye, C.O.; Gao, L.; Jiang, H.; Wu, Y.; Wang, Y.; Li, X.; Jiang, J. Whole-genome sequence analysis reveals phenanthrene and pyrene degradation pathways in newly isolated bacteria Klebsiella michiganensis EF4 and Klebsiella oxytoca ETN19. Microbiol. Res. 2023, 273, 127410. [Google Scholar] [CrossRef]
- Louvado, A.; Gomes, N.C.M.; Simões, M.M.Q.; Almeida, A.; Cleary, D.F.R.; Cunha, A. Polycyclic aromatic hydrocarbons in deep sea sediments: Microbe-pollutant interactions in a remote environment. Sci. Total Environ. 2015, 526, 312–328. [Google Scholar] [CrossRef]
- Bukowska, B.; Mokra, K.; Michalowicz, J. Benzo[a]pyrene—Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity. Int. J. Mol. Sci. 2022, 23, 6348. [Google Scholar] [CrossRef]
- Wang, Y.F.; Tam, N.F.Y. Microbial community dynamics and biodegradation of polycyclic aromatic hydrocarbons in polluted marine sediments in Hong Kong. Mar. Pollut. Bull. 2011, 63, 424–430. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Control FastQ | Results |
|---|---|
| High-quality readings | 10,613,744 (97.24%) |
| Readings due to low quality | 19.324 (0.18%) |
| Contained too much pollution | 336 reads (0.001539%) |
| Readings too short | 5.8520 (0.026808%) |
| Cut-out adapters | Yes |
| Properties and Characteristics | Total |
|---|---|
| Sequence size genome | 4,798,267 |
| No. of scaffolds | |
| Contigs >= 0 bps/1000 bps/50,000 bps | 51/28/12 |
| N50/L50 | 415,254/3 |
| No. of CDS | 4460 |
| No. of rRNA/tRNA/tmRNA | 2/77/1 |
| GC% | 54.38 |
| KEEG mapper reconstruction | |
| KEEG orthology (KO) | 2671 |
| Metabolism protein | 1487 |
| Genetic information processing | 690 |
| Signaling and cellular processes | 829 |
| Carbohydrate metabolism | 318 |
| Amino acid metabolism | 157 |
| Nucleotide metabolism | 104 |
| Metabolism of cofactors and vitamins | 134 |
| Energy metabolism | 101 |
| EggNOG-Mapper | |
| COG | 4349 |
| Pfam | 4186 |
| GO | 2540 |
| CAZy | 77 |
| BIGG | 1064 |
| Genomes NCBI | FAST ANI | ||||
|---|---|---|---|---|---|
| Strain | Genbank | Scientific Name | ANI Score | Fragment Length | Total Fragment |
| Crenshaw | GCA_016027695.1 | E. asburiae | 87.8055 | 1250 | 1586 |
| ATCC 35953 | GCA_001521715.1 | E. asburiae | 87.8536 | 1241 | 1586 |
| JM-458T.1 | GCA_900180435.1 | E. asburiae | 87.8124 | 1282 | 1586 |
| E1 | GCA_008364625.1 | E. dykesii | 87.8021 | 1262 | 1586 |
| DSM 16690 | GCA_001729805.1 | E. roggenkampii | 87.6385 | 1258 | 1586 |
| CCA6 | GCA_009176645.1 | E. oligotrophicus | 87.547 | 1218 | 1586 |
| WCHECL1597 | GCA_002939185.1 | E. sichuanensis | 87.326 | 1248 | 1586 |
| WCHECl-C4 | GCA_001984825.2 | E. chengduensis | 87.3321 | 1295 | 1586 |
| DSM-13645 | GCA_001729765.1 | E. kobei | 87.1635 | 1222 | 1586 |
| FDAARGOS 1428 | GCA_019047785.1 | E. cancerogenus | 86.5672 | 1214 | 1586 |
| EB-247 | GCA_900324475.1 | E. bugandensis | 87.4329 | 1290 | 1586 |
| ATCC BAA-2102 | GCA_001654845.1 | E. soli | 86.5295 | 1222 | 1586 |
| ATCC 23216 | GCA_000735515.1 | Leclercia adecarboxylata | 83.6121 | 1043 | 1586 |
| SB6411 | GCA_902158555.1 | Klebsiella spallanzanii | 80.9556 | 840 | 1586 |
| ATCC 8090 | GCA_011064845.1 | Citrobacter freundii | 81.5917 | 820 | 1586 |
| Modeling Server | Protein Modeling | Errat | Error/Warning/Plass | R. Plot% | Z-Score | SignalP | Num. aa | pI | Mol Weight | GRAVY |
|---|---|---|---|---|---|---|---|---|---|---|
| trRosseta | hpcB | 97.87 | 2/4/3 | 95.4% | −11.36 | No | 283 | 5.72 | 31721.04 | −0.228 |
| trRosseta | hpcC | 93.38 | 2/4/3 | 94.0% | −11.41 | No | 488 | 6.25 | 53130.86 | −0.112 |
| trRosseta | hpcD | 83.49 | 1/3/4 | 94.6% | −4.79 | No | 126 | 5.82 | 14336.35 | −0.202 |
| trRosseta | hpcE | 92.44 | 3/2/4 | 90.6% | −9.65 | No | 425 | 5.03 | 46227.37 | −0.146 |
| trRosseta | hpcG | 93.44 | 0/4/4 | 91.8% | −6.87 | No | 267 | 5.69 | 29481.68 | −0.081 |
| trRosseta | hpcH | 96.85 | 0/4/5 | 94.7% | −8.81 | No | 265 | 5.73 | 28175.33 | 0.107 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo, G.; Contreras-Liza, S.E.; Arbizu, C.I.; Rodriguez-Grados, P.M. Genome Sequencing Reveals the Potential of Enterobacter sp. Strain UNJFSC003 for Hydrocarbon Bioremediation. Genes 2025, 16, 89. https://doi.org/10.3390/genes16010089
Castillo G, Contreras-Liza SE, Arbizu CI, Rodriguez-Grados PM. Genome Sequencing Reveals the Potential of Enterobacter sp. Strain UNJFSC003 for Hydrocarbon Bioremediation. Genes. 2025; 16(1):89. https://doi.org/10.3390/genes16010089
Chicago/Turabian StyleCastillo, Gianmarco, Sergio Eduardo Contreras-Liza, Carlos I. Arbizu, and Pedro Manuel Rodriguez-Grados. 2025. "Genome Sequencing Reveals the Potential of Enterobacter sp. Strain UNJFSC003 for Hydrocarbon Bioremediation" Genes 16, no. 1: 89. https://doi.org/10.3390/genes16010089
APA StyleCastillo, G., Contreras-Liza, S. E., Arbizu, C. I., & Rodriguez-Grados, P. M. (2025). Genome Sequencing Reveals the Potential of Enterobacter sp. Strain UNJFSC003 for Hydrocarbon Bioremediation. Genes, 16(1), 89. https://doi.org/10.3390/genes16010089