
22q11.21 Deletions: A Review on the Interval Mediated by Low-Copy Repeats C and D

 , ,
, ,  and
and
Abstract
1. Introduction
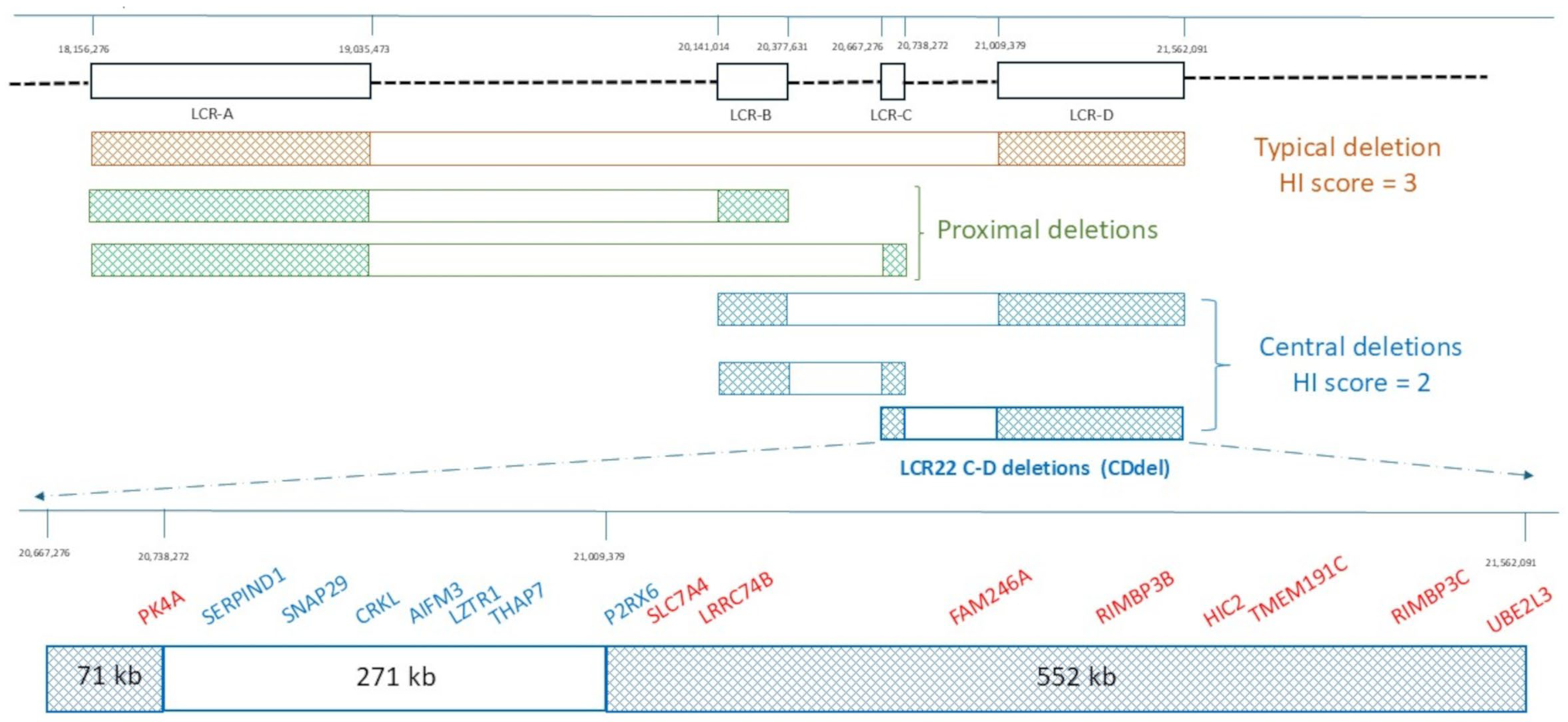
2. Architecture of the 22q11.2 Region
3. Gene Content
4. Long-Distance Effects of CNVs
5. CDdel Cases from Published Literature and Public Databases
5.1. More Frequent Pathological Traits
5.1.1. Renal/Urinary Tract Abnormalities
5.1.2. Cardiac Defects
5.1.3. Neurological/Behavioral Disorders
5.2. Less Frequent Pathological Traits
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, L.; Vermeesch, J.R. The 22q11.2 Low Copy Repeats. Genes 2022, 13, 2101. [Google Scholar] [CrossRef] [PubMed]
- Szczawińska-Popłonyk, A.; Schwartzmann, E.; Chmara, Z.; Głukowska, A.; Krysa, T.; Majchrzycki, M.; Olejnicki, M.; Ostrowska, P.; Babik, J. Chromosome 22q11.2 Deletion Syndrome: A Comprehensive Review of Molecular Genetics in the Context of Multidisciplinary Clinical Approach. Int. J. Mol. Sci. 2023, 24, 8317. [Google Scholar] [CrossRef] [PubMed]
- Bertini, V.; Azzarà, A.; Legitimo, A.; Milone, R.; Battini, R.; Consolini, R.; Valetto, A. Deletion Extents Are Not the Cause of Clinical Variability in 22q11.2 Deletion Syndrome: Does the Interaction between DGCR8 and miRNA-CNVs Play a Major Role? Front. Genet. 2017, 8, 47. [Google Scholar] [CrossRef]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef]
- Motta, M.; Fidan, M.; Bellacchio, E.; Pantaleoni, F.; Schneider-Heieck, K.; Coppola, S.; Borck, G.; Salviati, L.; Zenker, M.; Cirstea, I.C.; et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 2019, 28, 1007–1022. [Google Scholar] [CrossRef]
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11. 2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef]
- Racedo, S.E.; McDonald-McGinn, D.M.; Chung, J.H.; Goldmuntz, E.; Zackai, E.; Emanuel, B.S.; Zhou, B.; Funke, B.; Morrow, B.E. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am. J. Hum. Genet. 2015, 96, 235–244. [Google Scholar] [CrossRef]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Lyda, S.F.; Lei, E.P. Chromatin insulator mechanisms ensure accurate gene expression by controlling overall 3D genome organization. Curr. Opin. Genet. Dev. 2024, 87, 102208. [Google Scholar] [CrossRef] [PubMed]
- Rooney, K.; Levy, M.A.; Haghshenas, S.; Kerkhof, J.; Rogaia, D.; Tedesco, M.G.; Imperatore, V.; Mencarelli, A.; Squeo, G.M.; Di Venere, E.; et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. Int. J. Mol. Sci. 2021, 22, 8611. [Google Scholar] [CrossRef] [PubMed]
- Jalali, G.R.; Vorstman, J.A.; Errami, A.; Vijzelaar, R.; Biegel, J.; Shaikh, T.; Emanuel, B.S. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum. Mutat. 2008, 29, 433–440. [Google Scholar] [CrossRef]
- Ogilvie, C.M.; Ahnn, J.W.; Mann, K.; Roberts, R.G.; Flinter, F. A novel deletion in proximal 22q associated with cardiac septal defects and microcephaly: A case report. Mol. Cytogenet. 2009, 2, 9. [Google Scholar] [CrossRef]
- Fernández, L.; Nevado, J.; Santos, F.; Heine-Suñer, D.; Martinez-Glez, V.; García-Miñaur, S.; Palomo, R.; Delicado, A.; Pajares, I.L.; Palomares, M.; et al. A deletion and a duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Med. Genet. 2009, 10, 48. [Google Scholar] [CrossRef]
- Breckpot, J.; Thienpont, B.; Bauters, M.; Tranchevent, L.C.; Gewillig, M.; Allegaert, K.; Vermeesch, J.R.; Moreau, Y.; Devriendt, K. Congenital heart defects in a novel recurrent 22q11.2 deletion harboring the genes CRKL and MAPK1. Am. J. Med. Genet. A 2012, 158A, 574–580. [Google Scholar] [CrossRef]
- Garavelli, L.; Rosato, S.; Wischmeijer, A.; Gelmini, C.; Esposito, A.; Mazzanti, L.; Franchi, F.; De Crescenzo, A.; Palumbo, O.; Carella, M.; et al. 22q11.2 Distal Deletion Syndrome: Description of a New Case with Truncus Arteriosus Type 2 and Review. Mol. Syndr. 2011, 2, 35–44. [Google Scholar] [CrossRef]
- Zhao, W.; Niu, G.; Shen, B.; Zheng, Y.; Gong, F.; Wang, X.; Lee, J.; Mulvihill, J.J.; Chen, X.; Li, S. High-Resolution Analysis of Copy Number Variants in Adults with Simple-to-Moderate Congenital Heart Disease. Am. J. Med. Genet. Part A 2013, 161A, 3087–3094. [Google Scholar] [CrossRef]
- Rump, P.; de Leeuw, N.; van Essen, A.J.; Verschuuren-Bemelmans, C.C.; Veenstra-Knol, H.E.; Swinkels, M.E.; Oostdijk, W.; Ruivenkamp, C.; Reardon, W.; de Munnik, S.; et al. Central 22q11.2 deletions. Am. J. Med. Genet. A 2014, 164A, 2707–2723. [Google Scholar] [CrossRef]
- Burnside, R.D. 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet. Genome Res. 2015, 146, 89–99. [Google Scholar] [CrossRef]
- Kurahashi, H.; Nakayama, T.; Osugi, Y.; Tsuda, E.; Masuno, M.; Imaizumi, K.; Kamiya, T.; Sano, T.; Okada, S.; Nishisho, I. Deletion mapping of 22q11 in CATCH22 syndrome: Identification of a second critical region. Am. J. Hum. Genet. 1996, 58, 1377–1381. [Google Scholar] [PubMed]
- D’Angelo, C.S.; Jehee, F.S.; Koiffmann, C.P. An inherited atypical 1 Mb 22q11.2 deletion within the DGS/VCFS 3 Mb region in a child with obesity and aggressive behavior. Am. J. Med. Genet. A 2007, 143A, 1928–1932. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Graf, W.D.; Ramalingam, A.; Brawner, S.J.; Joyce, J.M.; Fiedler, S.; Zhou, X.G.; Liu, H.Y. Identification of Copy Number Variants on Human Chromosome 22 in Patients with a Variety of Clinical Findings. Cytogenet. Genome Res. 2011, 134, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Kiryluk, K.; Burgess, K.E.; Bodria, M.; Sampson, M.G.; Hadley, D.; Nees, S.N.; Verbitsky, M.; Perry, B.J.; Sterken, R.; et al. Copy-Number Disorders Are a Common Cause of Congenital Kidney Malformations. Am. J. Hum. Genet. 2012, 91, 987–997. [Google Scholar] [CrossRef]
- Verhagen, J.M.; Diderich, K.E.; Oudesluijs, G.; Mancini, G.M.; Eggink, A.J.; Verkleij-Hagoort, A.C.; Groenenberg, I.A.; Willems, P.J.; du Plessis, F.A.; de Man, S.A.; et al. Phenotypic variability of atypical 22q11.2 deletions not including TBX1. Am. J. Med. Genet. A 2012, 158A, 2412–2420. [Google Scholar] [CrossRef]
- Williams, C.L.; Nelson, K.R.; Grant, J.H.; Mikhail, F.M.; Robin, N.H. Cleft palate in a patient with the nested 22q11.2 LCR C to D deletion. Am. J. Med. Genet. A 2016, 170A, 260–262. [Google Scholar] [CrossRef]
- Clements, C.C.; Wenger, T.L.; Zoltowski, A.R.; Bertollo, J.R.; Miller, J.S.; de Marchena, A.B.; Mitteer, L.M.; Carey, J.C.; Yerys, B.E.; Zackai, E.H.; et al. Critical region within 22q11.2 linked to higher rate of autism spectrum disorder. Mol. Autism 2017, 8, 58. [Google Scholar] [CrossRef]
- Gavril, E.C.; Popescu, R.; Nucă, I.; Ciobanu, C.G.; Butnariu, L.I.; Rusu, C.; Pânzaru, M.C. Different Types of Deletions Created by Low-Copy Repeats Sequences Location in 22q11.2 Deletion Syndrome: Genotype-Phenotype Correlation. Genes 2022, 13, 2083. [Google Scholar] [CrossRef]
- Stefekova, A.; Capkova, P.; Capkova, Z.; Curtisova, V.; Srovnal, J.; Mracka, E.; Klaskova, E.; Prochazka, M. MLPA analysis of 32 foetuses with a congenital heart defect and 1 foetus with renal defects—Pilot study. The significant frequency rate of presented pathological CNV. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc Czech Repub. 2022, 166, 187–194. [Google Scholar] [CrossRef]
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. A 2018, 176, 2087–2098. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Bertini, V.; Milone, R.; Cristofani, P.; Cambi, F.; Bosetti, C.; Barbieri, F.; Bertelloni, S.; Cioni, G.; Valetto, A.; Battini, R. Enhancing DLG2 Implications in Neuropsychiatric Disorders: Analysis of a Cohort of Eight Patients with 11q14.1 Imbalances. Genes 2022, 12, 859. [Google Scholar] [CrossRef] [PubMed]
- Woodwar, K.J.; Stampalia, J.; Vanyai, H.; Rijhumal, H.; Potts, K.; Taylor, F.; Peverall, J.; Grumball, T.; Sivamoorthy, S.; Alinejad-Rokny, H.; et al. Atypical nested 22q11.2 duplications between LCR22B and LCR22D are associated with neurodevelopmental phenotypes including autism spectrum disorder with incomplete penetrance. Mol. Genet. Genomic. Med. 2019, 7, e00507. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Legitimo, A.; Bertini, V.; Alberio, A.M.Q.; Valetto, A.; Consolini, R. Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype. J. Clin. Med. 2023, 12, 7579. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Description | MORBID (Phenotype MIM Number) | HI Score | %HI | pLI |
|---|---|---|---|---|---|
| PI4KA *600286 | phosphatidylinositol 4-kinase alpha | AR #619708 Gastrointestinal defects and immunodeficiency syndrome AR #616531 Polymicrogyria, perisylvian, with cerebellar hypoplasia and arthrogryposis AR #619621 Spastic paraplegia | N | 35.17 | 0 |
| SERPIND1 *142360 | heparin cofactor II (serpin family D member1) | AD #612356 Thrombophilia 10 due to heparin cofactor II deficiency | N | 68.88 | 0 |
| SNAP29 *604202 | synaptosomal-associated protein, 29 kd | AR # 609528 Cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome (CEDNIK) | 30 | 61.36 | 0.01 |
| CRKL *602007 | CRK like proto-oncogene, adaptor protein | N | 1 | 4.35 | 0.45 |
| AIFM3 *617298 | apoptosis-inducing factor, mitochondrion-associated, 3 | N | N | 38.59 | 0 |
| LZTR1 *600574 | leucine zipper-like transcription regulator 1 | AD # 616564 Noonan syndrome 10 AR #605275 Noonan syndrome 2 AD #615670{Schwannomatosis-2, susceptibility to} | N | 32.82 | 0 |
| THAP7 *609518 | THAP domain- containing protein 7 | N | N | 50.87 | 0.01 |
| P2RX6 *608077 | purinergic receptor P2X-like 1 | N | N | 76.26 | 0 |
| SLC7A4 *603752 | solute carrier family 7, member 4 | N | N | 74.02 | 0 |
| LRRC74B N | leucine rich repeat containing 74B | N | N | N | 0 |
| FAM246A N | family with sequence similarity 246 member A | N | N | N | N |
| RIMBP3B *612700 | RIMS-binding protein 3B | N | N | 89.37 | 0.41 |
| HIC2 *607712 | hypermethylated in cancer 2 | N | N | 59.73 | 1 |
| TMEM191C N | transmembrane protein 191C | N | N | N | N |
| RIMBP3C *612701 | RIMS- binding protein 3C | N | N | 88.68 | N |
| UBE2L3 *603721 | ubiquitin-conjugating enzyme E2 L3 | N | N | 3.65 | 0.87 |
| Reference/ ID DECIPHER | Subjects | Sex/Age | Inheritance | Position (Mb) | Extent (kb) |
|---|---|---|---|---|---|
| Kurahashi et al., 1996 [21] | P1 | NR/NR | NR | NR | NR |
| D’Angelo et al., 2007 [22] | P2 P3: mother | F/8y F/NR | mat NR | NR | About 1000 |
| Yu et al., 2012 [23] | P4: case11 | M/NR | NR | 20.725–21.205 | 480 |
| P5: case12 | F/NR | NR | 20.725–21.205 | 480 | |
| P6: case13 | M/NR | NR | 20.725–21.205 | 480 | |
| Sanna-Cherchi et al., 2012 [24] | P7: ITA_25 | inherited | NR | 370-410 | |
| P8: ITA-13 | inherited | NR | 370-410 | ||
| P9: Czec_1 | dn | NR | 370-410 | ||
| Verhagen et al., 2012 [25] | (family A) P10: case 1 | F/32y | NR | 20.654–21.274 | 620 |
| (family C) P11: case 4 P12: case 5 (mother) | F/TP F/33y | mat NR | 20.683–21.274 | 591 | |
| (family D) P13: case 6 | F/18y | dn | 20.706–21.107 | 401 | |
| (family E) P14: case 7 P15: case 8 | M/5y F/37y | mat dn | 20.706–21.107 | 401 | |
| Rump et al., 2014 [19] | (family 2) P16: case 4 P17: case 5 (mother) | M/7y F (47,XXX)/31y | mat NR | 20.711–21.107 | 396 |
| (family 4) P18: case 7 P19: case 8 (father) P20: case 9 (brother) P21: case 10 (brother) P22: case 11 (grandfather) | M/8y M/38y M/5y M/13y M/68y | pat pat pat pat NR | 20.699–21.151 | 452 | |
| (family 8) P23: case16 | M/12y | NR | 20.656–21.574 | 918 | |
| (family 10) P24: case18 | M/4y | mat * | 20.706–21.107 | 401 | |
| (family 12) P25: case 21 | M/11y | mat * | 20.691–21.105 | 414 | |
| Racedo et al., 2015 [8] | P26 | NR | NR | NR | NR |
| Williams et al., 2016 [26] | P27 | F/6 w | NR | 20.726–21.086 | 360 |
| Lopez- Rivera et al., 2017 [9] | P28: patient 5 | M/1m | NR | 20.695–21.115 | 420 |
| P29: patient 6 | M/TP | NR | 20.705–21.115 | 410 | |
| P30: patient 7 | F/16y | NR | 20.705–21.105 | 400 | |
| P31: patient 8 | F/9y | NR | 20.705–21.105 | 400 | |
| P32: patient 9 | M/birth | NR | 20.715–21.105 | 390 | |
| P33: patient 10 | M/birth | NR | 20.725–21.115 | 380 | |
| P34: patient 11 | F/21y | NR | 20.735–21.115 | 370 | |
| P35: 12164-A | M/13m | NR | 20.715–21.105 | 390 | |
| P36: 12283-A | M/1y | NR | 20.715–21.105 | 390 | |
| Clements et al., 2017 [27] | P37, P38, P39, P40 (PX) | 3M-1F/ 2P <3y; 2P 15+y | NR | NR | NR |
| Gavril et al., 2022 [28] | P41 | F/14y | dn | NR | NR |
| Stefekova et al., 2022 [29] | P42 | NR/TP | NR | 20.695–21.111 | 416 |
| 249397 (DECIPHER) | P43 | F | inherited * | 20.678–21.585 | 907 |
| 249400 (DECIPHER) | P44 | F | NR | 20.721–21.095 | 374 |
| 251146 (DECIPHER) | P45 | M | NR | 20.706–21.107 | 401 |
| 255749 (DECIPHER) | P46 | M | inherited * | 20.740–21.109 | 368 |
| 262738 (DECIPHER) | P47 | F | NR | 20.721–21.025 | 304 |
| 273516 (DECIPHER) | P48 | M | inherited | 20.400–21.086 | 686 |
| 279514 (DECIPHER) | P49 | M | NR | 20.713–21.111 | 397 |
| 504011 (DECIPHER) | P50 | F | NR | 20.727–21.086 | 359 |
| 519448 (DECIPHER) | P51 | M | mat | 20.740–21.109 | 368 |
| 424297 (DECIPHER) | P52 | F | pat | 20.721–21.100 | 379 |
| 501528 (DECIPHER) | P53 | F | pat | 20.722–21.103 | 380 |
| 501567 (DECIPHER) | P54 | M | NR | 20.722–21.103 | 380 |
| 339858 (DECIPHER) | P55 | F | mat | 20.721–21.109 | 388 |
| 390043 (DECIPHER) | P56 | F | pat | 20.721–21.109 | 388 |
| Macro-Areas | Frequency | Excluded in N Cases | Anomaly Description |
|---|---|---|---|
| MORE FREQUENT | |||
| Renal/urinary abnormalities | 31.1% (23/74) 65 + 9 = 74 | N = 21 | mono/bilateral renal agenesis (P16, P29, P33, P34, P35, PX, P42, P47, P54); renal hypodysplasia (P7, P8, P9, P28, P31, P32); hydronephrosis (P18, P19, P36, P52); vesicoureteral reflux (P30, P32, P33); renal cyst/s (P10, P22, P28); uretero-pelvic junction stenosis (P20, P32) pyelonephritis (P20); megaureter (P28); renal hyperchogenicity (P36). |
| Cardiac defects | 14.5% (10/69) 65 + 4 = 69 | N = 21 | tetralogy of Fallot (P1, P10, P26, P36, PX); ventricular septal defect (P41, P51); atrio-ventricular septal defect (P17); pulmonary atresia (P1), pulmonary artery stenosis (P36), subpulmonary stenosis (P51), mitral valve incompetence (P17), dilated cardiomyopathy (P49), supraventricular tachycardia (P49), ventricular extra systole (P22). |
| Neurological/behavioral disorders | 52.1% (37/71) 65 + 6 = 71 | ||
| DD/ID | 38% (27/71) 27/65 + 6 | N = 8 | DD/ID (P4, P21, P54, P46, P48, nssv3466483, nssv3482339, nssv3472863, nssv3482015, nssv13648688, nssv580067); mild DD/ID (P10, P12, P17, P18, P25); severe DD/ID (P13, P14, P24); speech delay (P2, P5, P20, P35, P46); dyslexia (P18, P21, P23); learning disability (P2, P6, P19); motor delay (P15, P20, P25). |
| ASD | 4.2% (3/71) | ASD(P25); PDD (P14); PDD-NOS (P21, P25). | |
| ADHD spectrum disorder | 8.5% (6/71) | ADHD (PX); hyperactivity (P2, P43, P44, P48); ADD (P18); short attention span (P43). | |
| Behavioral and mood disorders | 7% (5/71) | N = 9 | psychosis (P43); aggressive and self-injurious behavior (P2, P18); hyperphagia, sleep problems (P2); OCD (PX); ODD (PX); depressive disorder (P3, PX); anxiety (P3, P18, PX). |
| Movement disorders | 9.9% (7/71) | stereotypic movements (P13); chorea (P13, P25); limb ataxia (P13); clumsiness (P17, P18, P19, P21); tremor (P17, P18, P19, P22); cramps (P17). | |
| “heterogeneous” neurological signs | 16.9% (12/71) | hypotonia (P14, P18, P21, P25, P47); joint hypermobility (P13, P51, P56); hyperreflexia (P15, P17, P18); Babinski reflexes (P17); mild ptosis (P14, P15, P18); dysphagia (P36); sensorineural hearing impairment (P50). | |
| LESS FREQUENT | |||
| Cleft palate/high arched palate | 9.2% (6/65) | N = 12 | high arched palate (P2, P3); high narrow palate (P18, P19, P22); bilateral cleft lip and palate (P27). |
| Genital anomalies | 10.8% (7/65) | N = 9 | mono/bilateral cryptorchidism (P14, P16, P28, P33); phymosis (P20, P32); epididymis and ductus deferens agenesis (P16); uterus bicornis (P10). |
| Skeletal anomalies | - | N = 13 | short stature (P12, P13, P23, P43, P45, P52); microcephaly (P5, P13, P27, P47); kyphosis (P17); scoliosis (P24); 6 lumbar vertebrae (PX); pes planus (P2); pes cavus (P17); 6 thoracic ribs (PX); missing canine (P23); hammered toes (P17); small hands (P2); craniosynostosis, ridged cranial sutures (P47); delay in bone (P23). |
| Morbidity | - | ear infections (P2, P18); recurrent infections (P14, PX, PX); inadequate vaccine response (PX, PX); low Ig (PX, PX). | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, V.; Cambi, F.; Legitimo, A.; Costagliola, G.; Consolini, R.; Valetto, A. 22q11.21 Deletions: A Review on the Interval Mediated by Low-Copy Repeats C and D. Genes 2025, 16, 72. https://doi.org/10.3390/genes16010072
Bertini V, Cambi F, Legitimo A, Costagliola G, Consolini R, Valetto A. 22q11.21 Deletions: A Review on the Interval Mediated by Low-Copy Repeats C and D. Genes. 2025; 16(1):72. https://doi.org/10.3390/genes16010072
Chicago/Turabian StyleBertini, Veronica, Francesca Cambi, Annalisa Legitimo, Giorgio Costagliola, Rita Consolini, and Angelo Valetto. 2025. "22q11.21 Deletions: A Review on the Interval Mediated by Low-Copy Repeats C and D" Genes 16, no. 1: 72. https://doi.org/10.3390/genes16010072
APA StyleBertini, V., Cambi, F., Legitimo, A., Costagliola, G., Consolini, R., & Valetto, A. (2025). 22q11.21 Deletions: A Review on the Interval Mediated by Low-Copy Repeats C and D. Genes, 16(1), 72. https://doi.org/10.3390/genes16010072