
Pan-Cancer Upregulation of the FOXM1 Transcription Factor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Analysis
2.2. Bulk RNA-Seq
2.3. Single-Cell RNA-Seq
3. Results
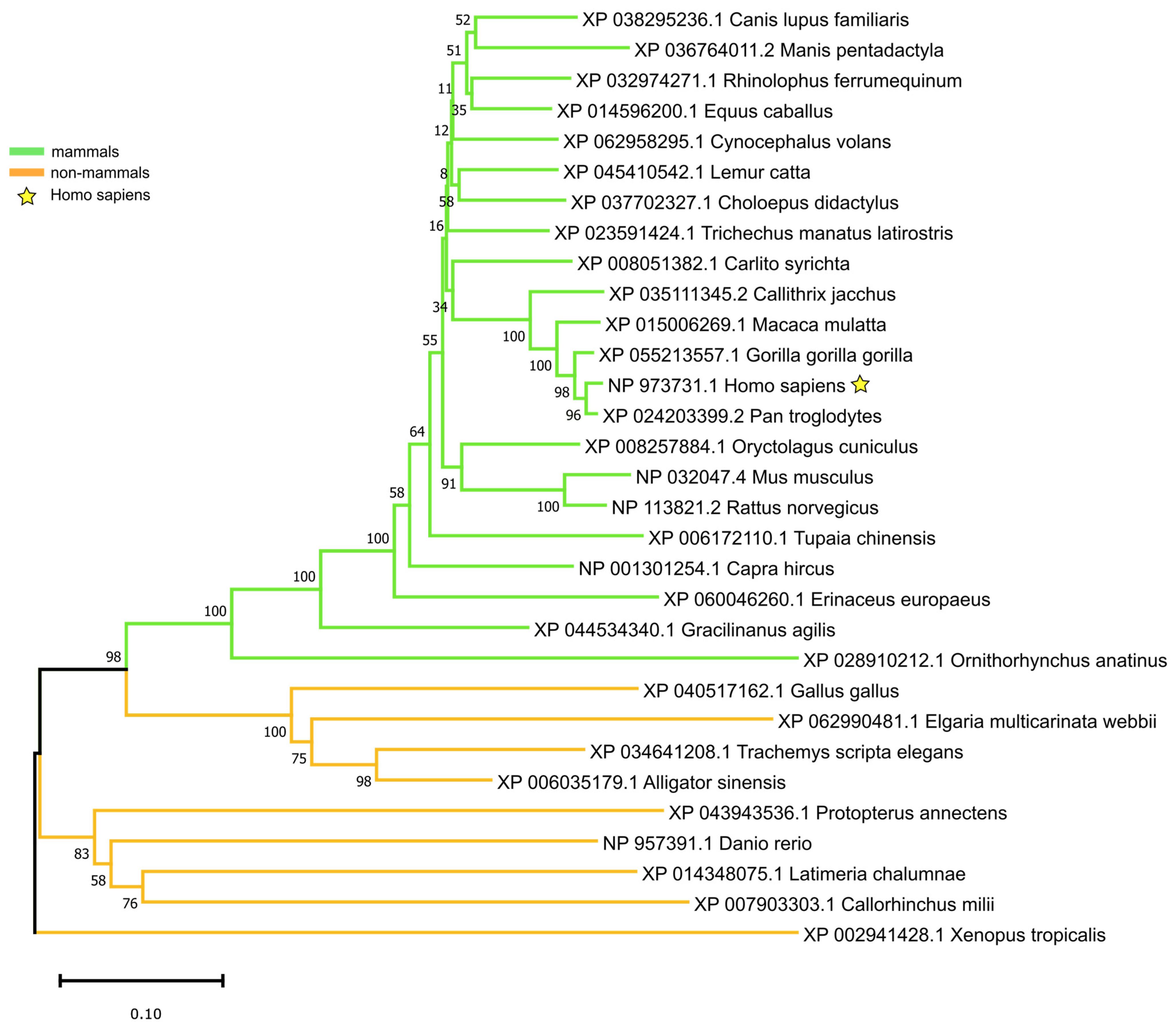
3.1. FOXM1 Sequence Across Species
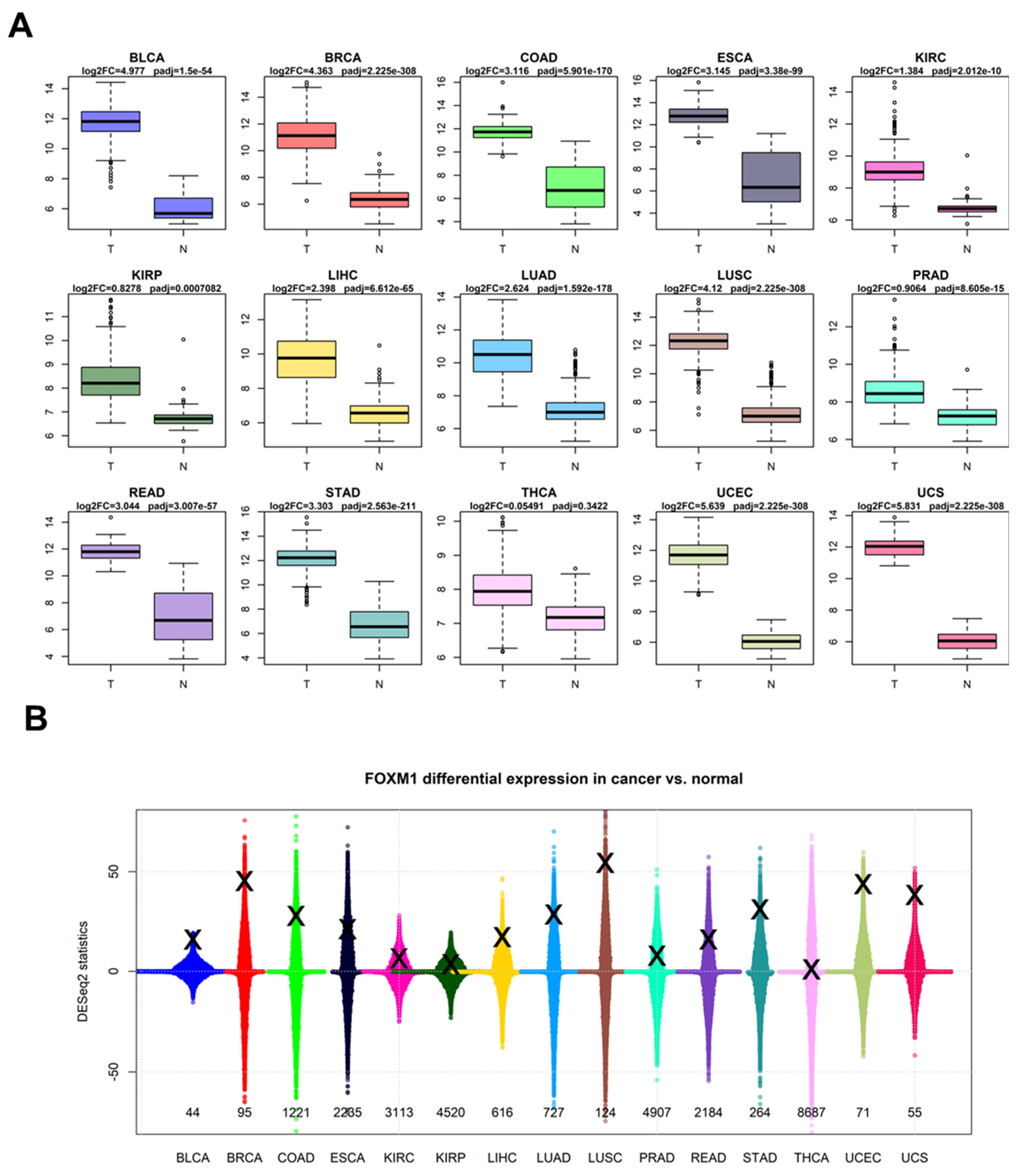
3.2. FOXM1 Expression in Bulk Cancer RNA-Seq
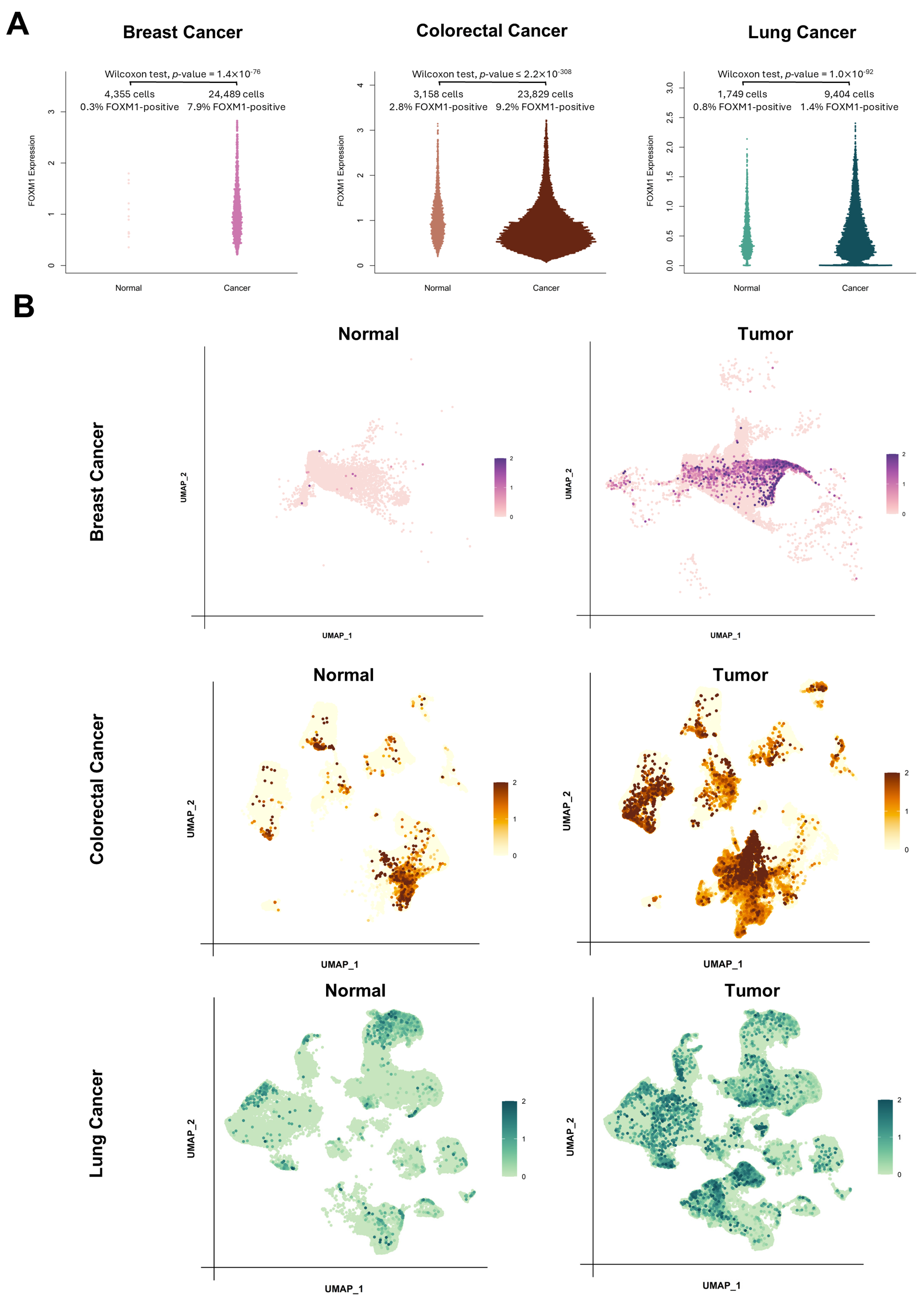
3.3. FOXM1 Expression in the Cancer Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Defilippo, A.; Giorgi, F.M.; Veltri, P.; Guzzi, P.H. Understanding Complex Systems through Differential Causal Networks. Sci. Rep. 2024, 14, 27431. [Google Scholar] [CrossRef] [PubMed]
- Torke, S.; Walther, W.; Stein, U. Immune Response and Metastasis—Links between the Metastasis Driver MACC1 and Cancer Immune Escape Strategies. Cancers 2024, 16, 1330. [Google Scholar] [CrossRef]
- Koo, C.-Y.; Muir, K.W.; Lam, E.W.-F. FOXM1: From Cancer Initiation to Progression and Treatment. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hannenhalli, S.; Kaestner, K.H. The Evolution of Fox Genes and Their Role in Development and Disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead Transcription Factors: Key Players in Health and Disease. Trends Genet. 2011, 27, 224–232. [Google Scholar] [CrossRef]
- Wierstra, I. Chapter Three—The Transcription Factor FOXM1 (Forkhead Box M1): Proliferation-Specific Expression, Transcription Factor Function, Target Genes, Mouse Models, and Normal Biological Roles**The Present Chapter Is Part I of a Two-Part Review on the Transcription Factor FOXM1. Part II of This FOXM1 Review Is Published in Volume 119 of Advances in Cancer Research: Inken Wierstra, FOXM1 (Forkhead Box M1) in Tumorigenesis: Overexpression in Human Cancer, Implication in Tumorigenesis, Oncogenic Functions, Tumor-Suppressive Properties and Target of Anti-Cancer Therapy. Advances in Cancer Research, 2013, Volume 119, in Press. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 118, pp. 97–398. [Google Scholar]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.T.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.R.; Medema, R.H. Activation of FoxM1 during G2 Requires Cyclin A/Cdk-Dependent Relief of Autorepression by the FoxM1 N-Terminal Domain. Mol. Cell. Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. Chapter Six—FOXM1 (Forkhead Box M1) in Tumorigenesis: Overexpression in Human Cancer, Implication in Tumorigenesis, Oncogenic Functions, Tumor-Suppressive Properties, and Target of Anticancer Therapy☆☆The Present Chapter Is Part II of a Two-Part Review on the Transcription Factor FOXM1. Part I of This FOXM1 Review Is Published in Volume 118 of Advances in Cancer Research: Inken Wierstra. The Transcription Factor FOXM1 (Forkhead Box M1): Proliferation-Specific Expression, Transcription Factor Function, Target Genes, Mouse Models, and Normal Biological Roles. Advances in Cancer Research, 2013, Volume 118, Pages 97–398. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 119, pp. 191–419. [Google Scholar]
- Myatt, S.S.; Lam, E.W.-F. The Emerging Roles of Forkhead Box (Fox) Proteins in Cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Bolte, C.; Zhang, Y.; Wang, I.-C.; Kalin, T.V.; Molkentin, J.D.; Kalinichenko, V.V. Expression of Foxm1 Transcription Factor in Cardiomyocytes Is Required for Myocardial Development. PLoS ONE 2011, 6, e22217. [Google Scholar] [CrossRef]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple Faces of FoxM1 Transcription Factor: Lessons from Transgenic Mouse Models. Cell Cycle 2011, 10, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Brison, C.M.; Nerli, S.; Arsenault, H.E.; McShan, A.C.; Chen, E.; Lee, H.-W.; Benanti, J.A.; Sgourakis, N.G.; Rubin, S.M. An Order-to-Disorder Structural Switch Activates the FoxM1 Transcription Factor. eLife 2019, 8, e46131. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, B.S.; Guillen, V.S.; Katzenellenbogen, J.A. Targeting the Oncogenic Transcription Factor FOXM1 to Improve Outcomes in All Subtypes of Breast Cancer. Breast Cancer Res. 2023, 25, 76. [Google Scholar] [CrossRef] [PubMed]
- Kopanja, D.; Chand, V.; O’Brien, E.; Mukhopadhyay, N.K.; Zappia, M.P.; Islam, A.B.M.M.K.; Frolov, M.V.; Merrill, B.J.; Raychaudhuri, P. Transcriptional Repression by FoxM1 Suppresses Tumor Differentiation and Promotes Metastasis of Breast Cancer. Cancer Res. 2022, 82, 2458–2471. [Google Scholar] [CrossRef]
- Wang, I.-C.; Ustiyan, V.; Zhang, Y.; Cai, Y.; Kalin, T.V.; Kalinichenko, V.V. Foxm1 Transcription Factor Is Required for the Initiation of Lung Tumorigenesis by Oncogenic KrasG12D. Oncogene 2014, 33, 5391–5396. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing Genetic Dysfunction across All Human Cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Diamant, I.; Clarke, D.J.B.; Evangelista, J.E.; Lingam, N.; Ma’ayan, A. Harmonizome 3.0: Integrated Knowledge about Genes and Proteins from Diverse Multi-Omics Resources. Nucleic Acids Res. 2024, gkae1080. [Google Scholar] [CrossRef] [PubMed]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 Is Required for Execution of the Mitotic Programme and Chromosome Stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W.-F. FOXM1: An Emerging Master Regulator of DNA Damage Response and Genotoxic Agent Resistance. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2014, 1839, 1316–1322. [Google Scholar] [CrossRef]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic Value of FOXM1 in Solid Tumors: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef] [PubMed]
- Paull, E.O.; Aytes, A.; Jones, S.J.; Subramaniam, P.S.; Giorgi, F.M.; Douglass, E.F.; Tagore, S.; Chu, B.; Vasciaveo, A.; Zheng, S.; et al. A Modular Master Regulator Landscape Controls Cancer Transcriptional Identity. Cell 2021, 184, 334–351.e20. [Google Scholar] [CrossRef]
- Aytes, A.; Mitrofanova, A.; Lefebvre, C.; Alvarez, M.J.; Castillo-Martin, M.; Zheng, T.; Eastham, J.A.; Gopalan, A.; Pienta, K.J.; Shen, M.M.; et al. Cross-Species Analysis of Genome-Wide Regulatory Networks Identifies a Synergistic Interaction between FOXM1 and CENPF That Drives Prostate Cancer Malignancy. Cancer Cell 2014, 25, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C.; et al. A Human B-cell Interactome Identifies MYB and FOXM1 as Master Regulators of Proliferation in Germinal Centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Brägelmann, J.; Schultze, J.L.; Perner, S. Web-TCGA: An Online Platform for Integrated Analysis of Molecular Cancer Data Sets. BMC Bioinform. 2016, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Carithers, L.J.; Moore, H.M. The Genotype-Tissue Expression (GTEx) Project. Biopreserv. Biobank 2015, 13, 307–308. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Maglott, D.R. RefSeq and LocusLink: NCBI Gene-Centered Resources. Nucleic Acids Res. 2001, 29, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Filipek, S.; Vogel, H. PyMOL and Inkscape Bridge the Data and the Data Visualization. Structure 2016, 24, 2041–2042. [Google Scholar] [CrossRef]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying Cancer and Normal RNA Sequencing Data from Different Sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.M.; Ceraolo, C.; Mercatelli, D. The R Language: An Engine for Bioinformatics and Data Science. Life 2022, 12, 648. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Cabrelle, C.; Veltri, P.; Giorgi, F.M.; Guzzi, P.H. Detection of Pan-Cancer Surface Protein Biomarkers via a Network-Based Approach on Transcriptomics Data. Brief. Bioinform. 2022, 23, bbac400. [Google Scholar] [CrossRef]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. Corto: A Lightweight R Package for Gene Network Inference and Master Regulator Analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional Characterization of Somatic Mutations in Cancer Using Network-Based Inference of Protein Activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Li, A.; Barber, R.F. Multiple Testing with the Structure-Adaptive Benjamini–Hochberg Algorithm. J. R. Stat. Soc. Ser. B Stat. Methodol. 2019, 81, 45–74. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A Single-Cell and Spatially Resolved Atlas of Human Breast Cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially Organized Multicellular Immune Hubs in Human Colorectal Cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef] [PubMed]
- Salcher, S.; Sturm, G.; Horvath, L.; Untergasser, G.; Kuempers, C.; Fotakis, G.; Panizzolo, E.; Martowicz, A.; Trebo, M.; Pall, G.; et al. High-Resolution Single-Cell Atlas Reveals Diversity and Plasticity of Tissue-Resident Neutrophils in Non-Small Cell Lung Cancer. Cancer Cell 2022, 40, 1503–1520.e8. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Kenny, M.; Schoen, I. Violin SuperPlots: Visualizing Replicate Heterogeneity in Large Data Sets. Mol. Biol. Cell 2021, 32, 1333–1334. [Google Scholar] [CrossRef] [PubMed]
- Berglund, A.-C.; Sjölund, E.; Östlund, G.; Sonnhammer, E.L.L. InParanoid 6: Eukaryotic Ortholog Clusters with Inparalogs. Nucleic Acids Res. 2008, 36, D263–D266. [Google Scholar] [CrossRef]
- Suhre, K.; McCarthy, M.I.; Schwenk, J.M. Genetics Meets Proteomics: Perspectives for Large Population-Based Studies. Nat. Rev. Genet. 2021, 22, 19–37. [Google Scholar] [CrossRef]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 626836. [Google Scholar] [CrossRef]
- Beljan, S.; Dominko, K.; Talajić, A.; Hloušek-Kasun, A.; Škrobot Vidaček, N.; Herak Bosnar, M.; Vlahoviček, K.; Ćetković, H. Structure and Function of Cancer-Related Developmentally Regulated GTP-Binding Protein 1 (DRG1) Is Conserved between Sponges and Humans. Sci. Rep. 2022, 12, 11379. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.-B.; Liu, J.; Wang, S.; Ma, L.; Zhang, J.-F. Biological Role and Expression of Translationally Controlled Tumor Protein (TCTP) in Tumorigenesis and Development and Its Potential for Targeted Tumor Therapy. Cancer Cell Int. 2024, 24, 198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, W.; Yang, Z.; Hu, Z.; Li, J. Multi-Omics Pan-Cancer Analyses Identify MCM4 as a Promising Prognostic and Diagnostic Biomarker. Sci. Rep. 2024, 14, 6517. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, D.; Giorgi, F.M.; Capuani, F.; Ciliberto, A.; Ciccarelli, F.D. Low Duplicability and Network Fragility of Cancer Genes. Trends Genet. 2008, 24, 427–430. [Google Scholar] [CrossRef]
- Dumont, J.E.; Maenhaut, C.; Lamy, F. Control of Thyroid Cell Proliferation and Goitrogenesis. Trends Endocrinol. Metab. 1992, 3, 12–17. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Mangan, S.; Zaslaver, A.; Alon, U. The Coherent Feedforward Loop Serves as a Sign-Sensitive Delay Element in Transcription Networks. J. Mol. Biol. 2003, 334, 197–204. [Google Scholar] [CrossRef]
- Ng, C.; Weigelt, B.; Grigoriadis, A.; Reis-Filho, J.S. Prognostic Signatures in Breast Cancer: Correlation Does Not Imply Causation. Breast Cancer Res. 2012, 14, 313. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, L.; Zhao, S.; Ai, Z.; Guo, J.; Liu, W. FoxM1 Expression Is Significantly Associated with Cisplatin-Based Chemotherapy Resistance and Poor Prognosis in Advanced Non-Small Cell Lung Cancer Patients. Lung Cancer 2013, 79, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. Targeting FOXM1 in Cancer. Biochem. Pharmacol. 2013, 85, 644–652. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozzobon, D.; Bellezza, A.; Giorgi, F.M. Pan-Cancer Upregulation of the FOXM1 Transcription Factor. Genes 2025, 16, 56. https://doi.org/10.3390/genes16010056
Pozzobon D, Bellezza A, Giorgi FM. Pan-Cancer Upregulation of the FOXM1 Transcription Factor. Genes. 2025; 16(1):56. https://doi.org/10.3390/genes16010056
Chicago/Turabian StylePozzobon, Daniele, Arianna Bellezza, and Federico M. Giorgi. 2025. "Pan-Cancer Upregulation of the FOXM1 Transcription Factor" Genes 16, no. 1: 56. https://doi.org/10.3390/genes16010056
APA StylePozzobon, D., Bellezza, A., & Giorgi, F. M. (2025). Pan-Cancer Upregulation of the FOXM1 Transcription Factor. Genes, 16(1), 56. https://doi.org/10.3390/genes16010056