
GENet: A Graph-Based Model Leveraging Histone Marks and Transcription Factors for Enhanced Gene Expression Prediction


Abstract
1. Introduction
- We have developed a novel supervised model architecture that integrates TF binding sites and histone modification data, specifically H3K27ac marks. This integration is crucial for accurately predicting gene expression levels, harnessing both the regulatory and epigenetic landscapes.
- Our model uniquely applies GCNs to handle the classification task for each feature type. This choice uses the structural nature of genomic data, allowing the model to capture and utilize the complex relationships between different genomic features and their influence on gene expression.
- We construct weighted sample similarity networks using cosine similarity to quantify and utilize the relationships among samples. This network construction facilitates the effective handling of the spatial and functional relationships inherent in genomic data.
- GENet introduces a cross-feature discovery tensor that captures correlations between labels across different features. This innovative structure allows for the integration of insights across the genomic landscape, enhancing predictive accuracy.
- The culmination of our methodology involves transforming the discovery tensor into a vector that inputs into a regression model. This final step synthesizes all prior analyses to provide a comprehensive and refined prediction of gene expression levels.
2. Methodologies
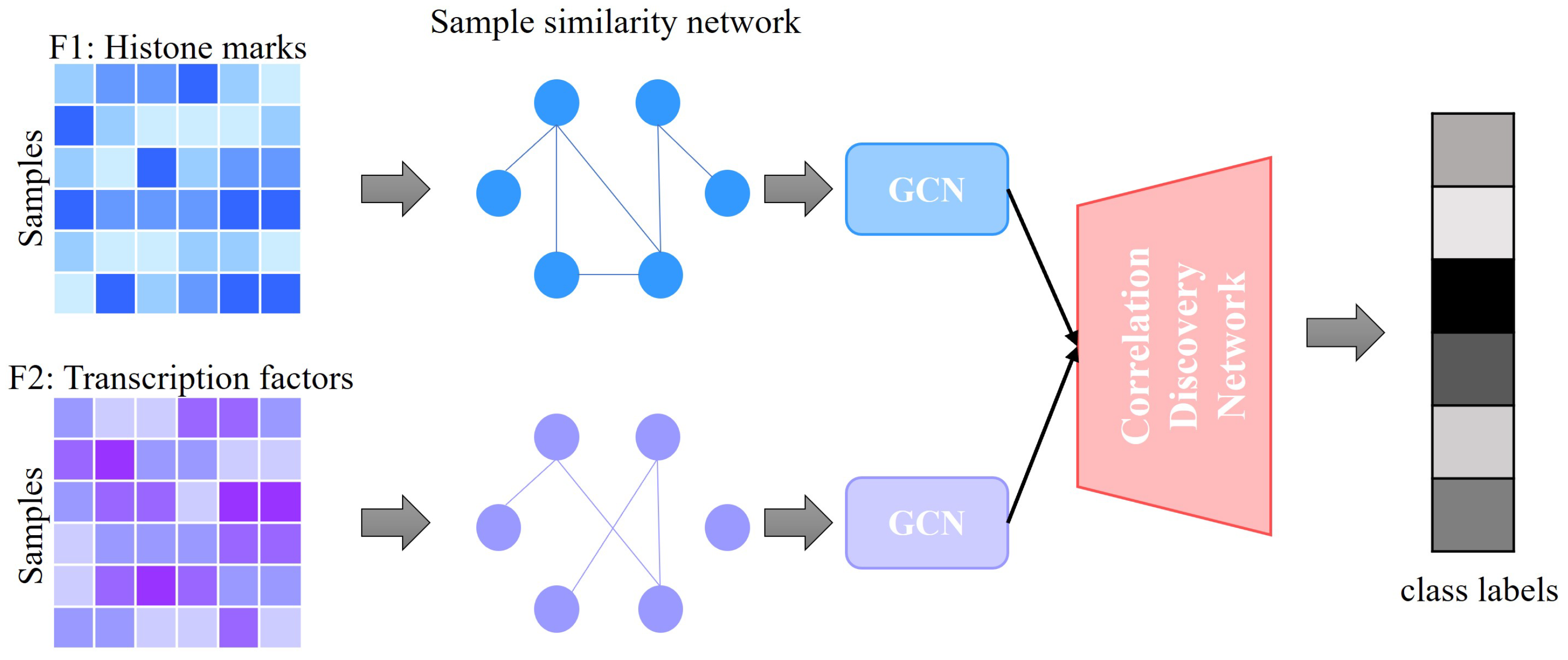
2.1. Framework of GENet
2.2. Feature Matrix Construction
| Algorithm 1: Construction of Cell Line- and Position-Specific Feature Matrices |
 |
2.3. Weighted Sample Similarity Networks
| Algorithm 2: Weighted sample similarity network construction |
 |
2.4. Training of Graph Convolutional Networks
2.5. Integration and Final Prediction
| Algorithm 3: GCN training and integration for gene expression prediction |
 |
3. Results and Discussion
3.1. Datasets
3.2. Baselines
- Linear Regression [21]: It served as our initial benchmark due to its simplicity and interpretability in modeling the relationship between independent variables and the target gene expression levels. This model provided a baseline for assessing the additional predictive value gained through more complex algorithms.
- Random Forest [22]: We utilized a random forest regressor configured with 100 decision trees to capture nonlinear relationships and interactions among features. This ensemble method is renowned for its performance in regression tasks, offering insights into the significance of using multiple learning models for improved predictions.
- GBM [23]: They were employed to further explore the potential of ensemble learning in enhancing predictive accuracy. The GBM model, consisting of 100 boosting stages, aimed to sequentially correct errors of weak learners, thereby strengthening the model’s ability to predict gene expression levels accurately.
- SVM [24]: SVM with a radial basis function (RBF) kernel was chosen for its capacity to handle both linear and nonlinear data structures. This model’s inclusion allowed us to explore the utility of margin maximization in the context of gene expression prediction.
- Simple Neural Network: To incorporate the advantages of deep learning, a simple neural network architecture comprising an input layer, a hidden layer with 64 units followed by a ReLU activation function, and an output layer was implemented. This model tested the hypothesis that deep learning techniques could capture complex, high-level abstractions from the genomic data.
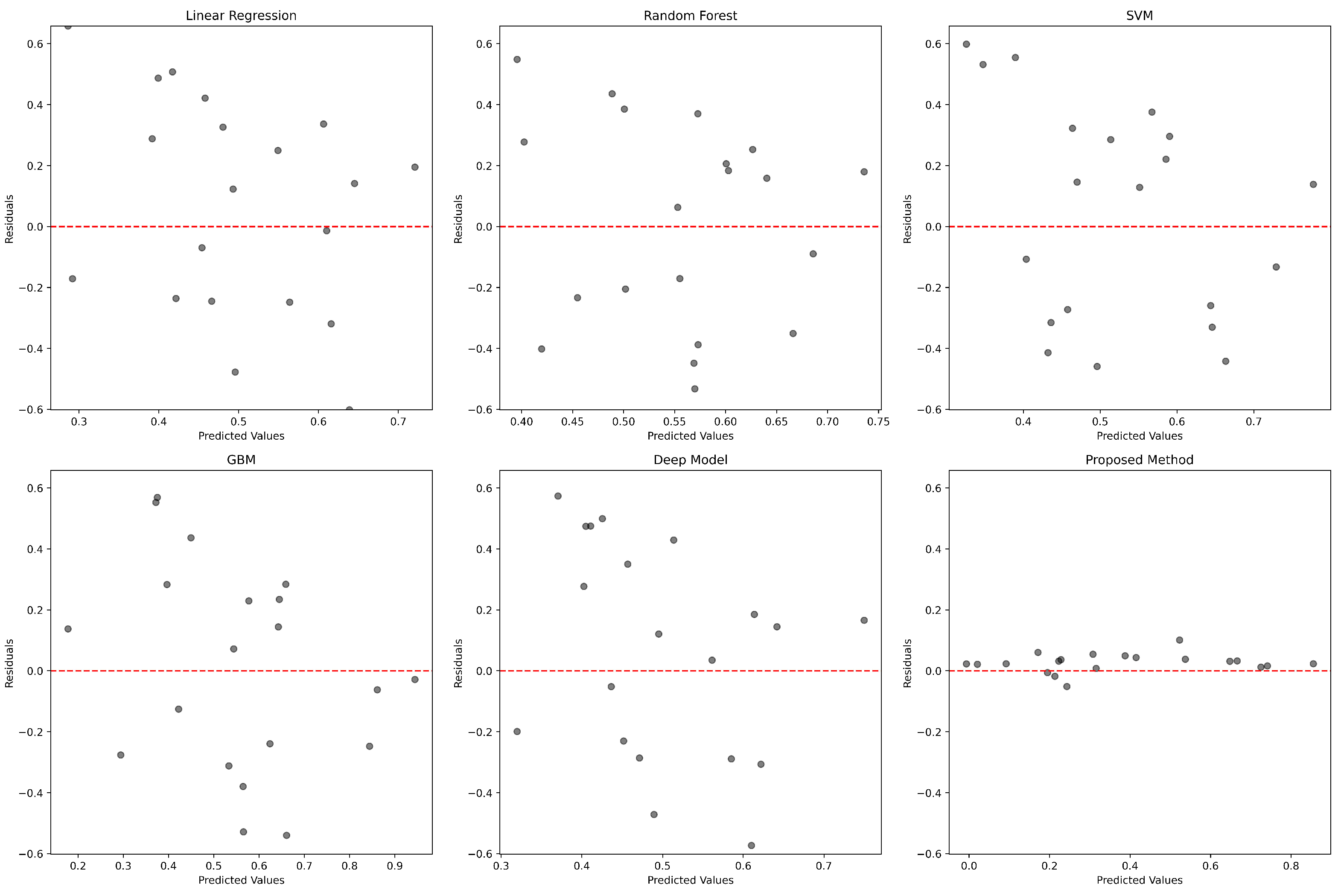
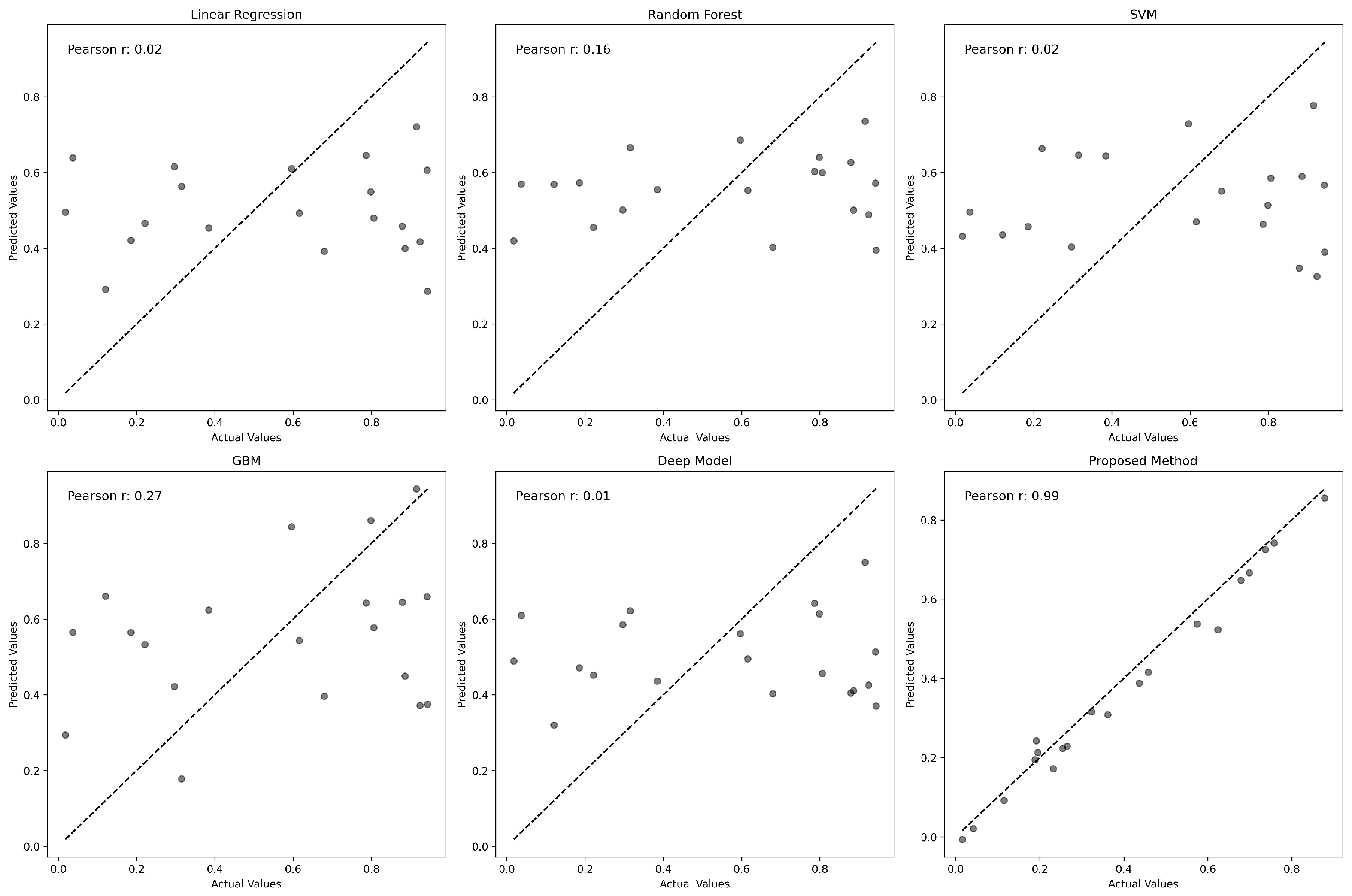
3.3. Comparative Performance Analysis of GENet and Other Predictive Models
3.4. Hyperparameter Tuning
4. Study Limitation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pascual-Ahuir, A.; Fita-Torró, J.; Proft, M. Capturing and understanding the dynamics and heterogeneity of gene expression in the living cell. Int. J. Mol. Sci. 2020, 21, 8278. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T. Regulation of transcription and gene expression in eukaryotes. Nat. Educ. 2008, 1, 199. [Google Scholar]
- Chen, C.H.; Zheng, R.; Tokheim, C.; Dong, X.; Fan, J.; Wan, C.; Tang, Q.; Brown, M.; Liu, J.S.; Meyer, C.A.; et al. Determinants of transcription factor regulatory range. Nat. Commun. 2020, 11, 2472. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Hardy, K.; Bunting, K.L.; Ma, L.; Peng, K.; Chen, X.; Shannon, M.F. Defining the chromatin signature of inducible genes in T cells. Genome Biol. 2009, 10, R107. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Greven, M.C.; Kundaje, A.; Djebali, S.; Brown, J.B.; Cheng, C.; Gingeras, T.R.; Gerstein, M.; Guigó, R.; Birney, E.; et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012, 13, R53. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Roider, H.G.; do Rego, T.G.; de Carvalho, F.d.T. Predicting gene expression in T cell differentiation from histone modifications and transcription factor binding affinities by linear mixture models. BMC Bioinform. 2011, 12, S29. [Google Scholar] [CrossRef] [PubMed]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Hassen, R.; Le, N. Combinatorial roles of dna methylation and histone modifications on gene expression. In Some Current Advanced Researches on Information and Computer Science in Vietnam: Post, Proceedings of the First NAFOSTED Conference on Information and Computer Science, Ha Noi, Vietnam, 13–14 March 2014; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Cheng, C.; Gerstein, M. Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells. Nucleic Acids Res. 2012, 40, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ching, T.; Huang, S.; Garmire, L.X. Using epigenomics data to predict gene expression in lung cancer. BMC Bioinform. 2015, 16, S10. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lanchantin, J.; Robins, G.; Qi, Y. DeepChrome: Deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016, 32, i639–i648. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lanchantin, J.; Robins, G.; Qi, Y. Attend and predict: Understanding gene regulation by selective attention on chromatin. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Volume 30. [Google Scholar]
- Sekhon, A.; Singh, R.; Qi, Y. DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications. Bioinformatics 2018, 34, i891–i900. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Theesfeld, C.L.; Yao, K.; Chen, K.M.; Wong, A.K.; Troyanskaya, O.G. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018, 50, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- McLeay, R.; Lesluyes, T.; Partida, G.C.; Bailey, T.L. Genome-wide in silico prediction of gene expression. Bioinformatics 2012, 28, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Gasparoni, N.; Gasparoni, G.; Gianmoena, K.; Cadenas, C.; Polansky, J.K.; Ebert, P.; Nordström, K.; Barann, M.; Sinha, A.; et al. Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res. 2017, 45, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Zhou, Q.; Wong, W. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 21521–21526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tong, H.; Xu, J.; Maciejewski, R. Graph convolutional networks: A comprehensive review. Comput. Soc. Netw. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S. Applied Linear Regression; John Wiley & Sons: Hoboken, NJ, USA, 2005; Volume 528. [Google Scholar]
- Biau, G.; Scornet, E. A random forest guided tour. Test 2016, 25, 197–227. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobotics 2013, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Suthaharan, S.; Suthaharan, S. Support vector machine. In Machine Learning Models and Algorithms for Big Data Classification: Thinking with Examples for Effective Learning; Springer: New York, NY, USA, 2016; pp. 207–235. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | MSE | RMSE | MAE | |
|---|---|---|---|---|
| Linear Regression | 0.1223 | 0.3498 | 0.3058 | 0.1519 |
| Random Forest | 0.1054 | 0.3247 | 0.2940 | 0.0075 |
| SVM | 0.1212 | 0.3482 | 0.3165 | 0.1415 |
| GBM | 0.1080 | 0.3287 | 0.2841 | 0.0173 |
| Deep Model | 0.1208 | 0.3475 | 0.3068 | 0.1374 |
| Proposed Method | 0.0334 | 0.1828 | 0.01295 | 0.9968 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labani, M.; Beheshti, A.; O’Brien, T.A. GENet: A Graph-Based Model Leveraging Histone Marks and Transcription Factors for Enhanced Gene Expression Prediction. Genes 2024, 15, 938. https://doi.org/10.3390/genes15070938
Labani M, Beheshti A, O’Brien TA. GENet: A Graph-Based Model Leveraging Histone Marks and Transcription Factors for Enhanced Gene Expression Prediction. Genes. 2024; 15(7):938. https://doi.org/10.3390/genes15070938
Chicago/Turabian StyleLabani, Mahdieh, Amin Beheshti, and Tracey A. O’Brien. 2024. "GENet: A Graph-Based Model Leveraging Histone Marks and Transcription Factors for Enhanced Gene Expression Prediction" Genes 15, no. 7: 938. https://doi.org/10.3390/genes15070938
APA StyleLabani, M., Beheshti, A., & O’Brien, T. A. (2024). GENet: A Graph-Based Model Leveraging Histone Marks and Transcription Factors for Enhanced Gene Expression Prediction. Genes, 15(7), 938. https://doi.org/10.3390/genes15070938