
Evaluation of Toll-like Receptor 4 (TLR4) Involvement in Human Atrial Fibrillation: A Computational Study

 ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Selection and Analysis
2.2. Functional Analysis of Differentially Expressed Genes (DEGs) and Assessment of TLR4 Involvement in AF
2.3. Statistical Analysis
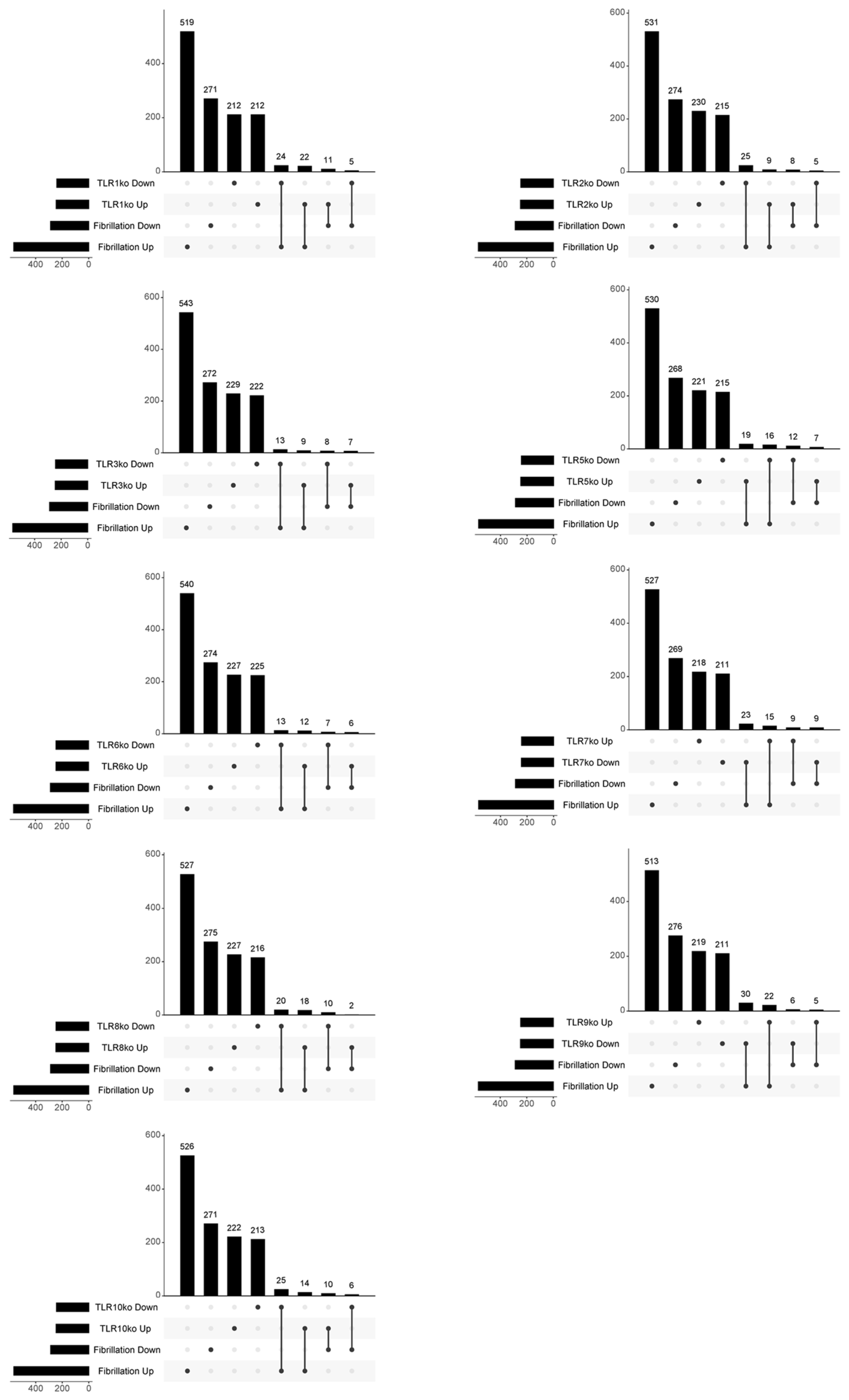
3. Results
3.1. Meta-Analysis Results and Pathway Enrichment Analysis
3.2. In-Depth Exploration of the Involvement of TLR4 Signalling in AF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steward, R.; McNally, F.J.; Schedl, P. Isolation of the Dorsal Locus of Drosophila. Nature 1984, 311, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Ryffel, B.; Quesniaux, V.F.J.; Cartwright, N.; Paul-Clark, M. Role of Pattern-Recognition Receptors in Cardiovascular Health and Disease. Biochem. Soc. Trans. 2007, 35, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Naito, S. Tissue-Specific mRNA Expression Profiles of Human Toll-like Receptors and Related Genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and Localization of Toll-like Receptor Signalling Complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Tetta, C.; Moula, A.I.; Matteucci, F.; Parise, O.; Maesen, B.; Johnson, D.; La Meir, M.; Gelsomino, S. Association between Atrial Fibrillation and Helicobacter Pylori. Clin. Res. Cardiol. 2019, 108, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, A.; Platek, A.E.; Dluzniewski, M.; Szymanski, F.M. History of Lyme Disease as a Predictor of Atrial Fibrillation. Am. J. Cardiol. 2020, 125, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Bendtzen, K. Are Microbiomes Involved in Paroxysmal Atrial Fibrillation? Repeated Clinical Improvement after Pivmecillinam (Amdinocillin) Therapy. Acad. Lett. 2021, 3124. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Y.; Bai, Y.; Luo, Q.; Lin, X.; Yang, Q.; Wang, S.; Xin, H. Gut Microbiota and Metabolites in Atrial Fibrillation Patients and Their Changes after Catheter Ablation. Microbiol. Spectr. 2022, 10, e0107721. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The Emerging Role of Toll-like Receptor 4 in Myocardial Inflammation. Cell Death Dis. 2016, 7, e2234. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Yusung, S.; Frisancho, S.; Barrett, M.; Gatewood, S.; Steele, R.; Rose, N.R. IL-12 Receptor Beta 1 and Toll-like Receptor 4 Increase IL-1 Beta- and IL-18-Associated Myocarditis and Coxsackievirus Replication. J. Immunol. 2003, 170, 4731–4737. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.; Leonard, F.; Ernens, I.; Rodius, S.; Vausort, M.; Rolland-Turner, M.; Devaux, Y.; Wagner, D.R. Adenosine Reduces Cell Surface Expression of Toll-like Receptor 4 and Inflammation in Response to Lipopolysaccharide and Matrix Products. J. Cardiovasc. Transl. Res. 2011, 4, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Inflammation in Cardiac Injury, Repair and Regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Tang, F.; Zhang, J.; Luan, A.; Mei, M.; Xu, C.; Zhang, S.; Wang, H.; Maslov, L.N. Astragaloside IV Attenuates Injury Caused by Myocardial Ischemia/Reperfusion in Rats via Regulation of Toll-like Receptor 4/Nuclear Factor-κB Signaling Pathway. Phytother. Res. 2015, 29, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, A.; Chong, A.J.; Yada, M.; Shomura, S.; Takayama, H.; Fleisig, A.J.; Agnew, M.L.; Hampton, C.R.; Rothnie, C.L.; Spring, D.J.; et al. Inhibition of Toll-like Receptor 4 with Eritoran Attenuates Myocardial Ischemia-Reperfusion Injury. Circulation 2006, 114, I270–I274. [Google Scholar] [CrossRef] [PubMed]
- Staerk, L.; Wang, B.; Preis, S.R.; Larson, M.G.; Lubitz, S.A.; Ellinor, P.T.; McManus, D.D.; Ko, D.; Weng, L.-C.; Lunetta, K.L.; et al. Lifetime Risk of Atrial Fibrillation According to Optimal, Borderline, or Elevated Levels of Risk Factors: Cohort Study Based on Longitudinal Data from the Framingham Heart Study. BMJ 2018, 361, k1453. [Google Scholar] [CrossRef] [PubMed]
- Piccini, J.P.; Hammill, B.G.; Sinner, M.F.; Jensen, P.N.; Hernandez, A.F.; Heckbert, S.R.; Benjamin, E.J.; Curtis, L.H. Incidence and Prevalence of Atrial Fibrillation and Associated Mortality among Medicare Beneficiaries, 1993–2007. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Friberg, L.; Bergfeldt, L. Atrial Fibrillation Prevalence Revisited. J. Intern. Med. 2013, 274, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Kwan, G.F.; Benjamin, E.J. Global Epidemiology of Atrial Fibrillation. Nat. Rev. Cardiol. 2014, 11, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Hart, C.L.; Hole, D.J.; McMurray, J.J.V. A Population-Based Study of the Long-Term Risks Associated with Atrial Fibrillation: 20-Year Follow-up of the Renfrew/Paisley Study. Am. J. Med. 2002, 113, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.-C.; Lin, Y.-C.; Chang, S.-H.; Chang, G.-J.; Hsu, Y.-J.; Lin, Y.-M.; Lee, Y.-S.; Wang, C.-L.; Yeh, Y.-H. Differential Left-to-Right Atria Gene Expression Ratio in Human Sinus Rhythm and Atrial Fibrillation: Implications for Arrhythmogenesis and Thrombogenesis. Int. J. Cardiol. 2016, 222, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Cabrera, C.P.; Finlay, M.; Lall, K.; Nobles, M.; Schilling, R.J.; Wood, K.; Mein, C.A.; Barnes, M.R.; Munroe, P.B.; et al. Differentially Expressed Genes for Atrial Fibrillation Identified by RNA Sequencing from Paired Human Left and Right Atrial Appendages. Physiol. Genom. 2019, 51, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Adam, O.; Lavall, D.; Theobald, K.; Hohl, M.; Grube, M.; Ameling, S.; Sussman, M.A.; Rosenkranz, S.; Kroemer, H.K.; Schäfers, H.-J.; et al. Rac1-Induced Connective Tissue Growth Factor Regulates Connexin 43 and N-Cadherin Expression in Atrial Fibrillation. J. Am. Coll. Cardiol. 2010, 55, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-H.; Kuo, C.-T.; Lee, Y.-S.; Lin, Y.-M.; Nattel, S.; Tsai, F.-C.; Chen, W.-J. Region-Specific Gene Expression Profiles in the Left Atria of Patients with Valvular Atrial Fibrillation. Heart Rhythm 2013, 10, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hunt, G.P.; Grassi, L.; Henkin, R.; Smeraldi, F.; Spargo, T.P.; Kabiljo, R.; Koks, S.; Ibrahim, Z.; Dobson, R.J.B.; Al-Chalabi, A.; et al. GEOexplorer: A Webserver for Gene Expression Analysis and Visualisation. Nucleic Acids Res. 2022, 50, W367–W374. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. Available online: https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/cpz1.90 (accessed on 9 February 2024). [CrossRef] [PubMed]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Koleti, A.; Terryn, R.; Stathias, V.; Chung, C.; Cooper, D.J.; Turner, J.P.; Vidovic, D.; Forlin, M.; Kelley, T.T.; D’Urso, A.; et al. Data Portal for the Library of Integrated Network-Based Cellular Signatures (LINCS) Program: Integrated Access to Diverse Large-Scale Cellular Perturbation Response Data. Nucleic Acids Res. 2018, 46, D558–D566. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering Workflow-Based Network Analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, X.; Bao, X.; Xiao, W.; Chen, G. Toll-like Receptor 4 (TLR4) Inhibitors: Current Research and Prospective. Eur. J. Med. Chem. 2022, 235, 114291. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. The Emerging Role of Innate Immunity in the Heart and Vascular System: For Whom the Cell Tolls. Circ. Res. 2011, 108, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Long, Y.; He, X.; Li, Y. Effects of different doses of glucocorticoids on postoperative atrial fibrillation: A meta-analysis. BMC Cardiovasc. Disord. 2023, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Tobón, C.; Palacio, L.C.; Chidipi, B.; Slough, D.P.; Tran, T.; Tran, N.; Reiser, M.; Lin, Y.S.; Herweg, B.; Sayad, D.; et al. The Antimalarial Chloroquine Reduces the Burden of Persistent Atrial Fibrillation. Front. Pharmacol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.J.; Li, S.J.; Lu, Y.Y.; Wu, W.S.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Toll-like receptor 4 activation modulates pericardium-myocardium interactions in lipopolysaccharide-induced atrial arrhythmogenesis. Europace 2021, 23, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Shields, K.J.; Manzi, S.; Wasko, M.C.; Sharma, T.S. Association of Hydroxychloroquine Use With Decreased Incident Atrial Fibrillation in Systemic Lupus Erythematosus. Arthritis Care Res. 2021, 73, 828–832. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.; Iacobellis, G.; Li, N.; Nattel, S.; Goldberger, J. Targeting the Substrate for Atrial Fibrillation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2024, 83, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting between-sample RNA-Seq normalizationmethods from the perspective of their assumptions. Brief. Bioinform. 2018, 19, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, H.A.; Bhattacharyya, D.K.; Kalita, J.K. Differential Expression Analysis of RNA-seq Reads: Overview, Taxonomy, and Tools. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 566–586. [Google Scholar] [CrossRef] [PubMed]
- Milind, N.; Preuss, C.; Haber, A.; Ananda, G.; Mukherjee, S.; John, C.; Shapley, S.; Logsdon, B.A.; Crane, P.K.; Carter, G.W. Transcriptomic stratification of late-onset Alzheimer’s cases reveals novel genetic modifiers of disease pathology. PLoS Genet. 2020, 16, e1008775. [Google Scholar] [CrossRef] [PubMed]
- Kiltschewskij, D.J.; Cairns, M.J. Transcriptome-Wide Analysis of Interplay between mRNA Stability, Translation and Small RNAs in Response to Neuronal Membrane Depolarization. Int. J. Mol. Sci. 2020, 21, 7086. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhou, Y.; Cai, Z.; Terekhova, M.; Swain, A.; Andhey, P.S.; Guimaraes, R.M.; Ulezko Antonova, A.; Qiu, T.; Sviben, S.; et al. Transcriptomic atlas and interaction networks of brain cells in mouse CNS demyelination and remyelination. Cell Rep. 2023, 42, 112293. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Djeddi, S.; Dunlap, G.; He, W.; Jia, X.; Burk, C.M.; Ryan, T.; McGill, A.; Allegretti, J.R.; Kataru, R.P.; et al. TCF1-LEF1 co-expression identifies a multipotent progenitor cell (TH2-MPP) across human allergic diseases. Nat. Immunol. 2024, 25, 902–915. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Term | Overlap | p-Value | Adj. p-Value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|
| Fibrillation Up | TLR4_ko Down | 17/243 | 0.001 | 0.001 | 2.637 | 19.7 | PHTF2;JUND;TACSTD2;CXCR4;HSPA2;NXT2;REXO2;ETS1;ZFP36L2;GULP1;PLAC8;RCAN1;MAPKAPK2;ANGPTL2;YPEL5;PDLIM5;S100A8 |
| TLR4_ko Up | 8/246 | 0.394 | 0.395 | 1.158 | 1.1 | LYN;NUP50;CCL5;GZMA;ID1;ARHGDIB;DNPH1;SNX5 | |
| Fibrillation Down | TLR4_ko Down | 6/243 | 0.138 | 0.215 | 1.755 | 3.5 | GRB7;TFAP2C;PERP;SOSTDC1;PHLDA2;PPL |
| TLR4_ko Up | 9/246 | 0.009 | 0.039 | 2.660 | 12.4 | GRAMD1C;C7;ANXA3;SCNN1A;NR2F1;SLC4A4;SORL1;EREG;DCBLD2 |
| Term | Overlap | p-Value | Adj. p-Value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|
| Fibrillation Up | MYD88_ko Down | 27/246 | 1.9 × 10−9 | 2.7 × 10−8 | 4.40 | 88.4 | CDS1;LST1;CFI;TACSTD2;ITPR1;HBB;TCF21;PLAC8;ALOX5;ARHGDIB;CASP1;HLA-DQA1;LYN;IRX5;CD300A;IGFBP2;RNASE6;LAPTM5;PPBP;MAPK14;NUDT21;TYROBP;SELL;MZB1;B3GNT2;FCGR2C;S100A8 |
| MYD88_ko Up | 20/243 | 2.1 × 10−5 | 5.5 × 10−5 | 3.16 | 34.1 | CFD;COL15A1;VWF;C1S;MTCL1;FHL1;FHL2;DPT;GULP1;AKAP12;ACTA1;MFAP4;EPB41L3;MAPKAPK2;COL5A2;TSPAN4;PDLIM5;CHGB;TGM2;FGFR1 | |
| TICAM1_ko Down | 15/245 | 4.3 × 10−3 | 5.5 × 10−3 | 2.28 | 12.4 | UBE3C;FHL2;CYBA;HNMT;MS4A4A;RHOB;MARCO;COL4A2;UST;MZB1;TXNIP;ANGPTL2;CRK;S100A8;CHGB | |
| TICAM1 _ko Up | 13/244 | 2.2 × 10−2 | 2.5 × 10−2 | 1.96 | 7.5 | CD74;PHTF2;ADAM10;CRIP2;SORBS2;HMGB1;GULP1;MFAP4;MECOM;CDH2;TCF4;SKP2;CRYM | |
| Fibrillation Down | MYD88_ko Down | 10/246 | 3.0 × 10−3 | 2.1 × 10−2 | 2.98 | 17.3 | CA12;PTPRZ1;FLRT3;CHL1;ANXA3;ANPEP;PLEKHB1;CHI3L1;SORL1;ST6GALNAC5 |
| Term | Overlap | p-Value | Adj. p-Value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|
| Fibrillation UP | TLR1_ko Down | 24/241 | 1 × 10−7 | 7 × 10−7 | 3.93 | 63.4 | BRD2;RAB1A;POSTN;COL15A1;DDX3X;LIMCH1;DUSP1;FHL2;HSPA2;PRSS23;GULP1;NFKBIA;MFAP4;MYO1B;MECOM;OAS1;CCNG2;EPB41L3;ID1;PDK4;PHACTR2;EIF4E2;SRSF10;CHGB |
| TLR1_ko Up | 22/245 | 2 × 10−6 | 8 × 10−6 | 3.49 | 45.9 | SRGN;ABCC4;RRM1;GYPC;GZMA;LST1;CFI;LAPTM5;TACSTD2;HBB;TCF21;CYBB;CRIP2;PPBP;SORBS1;PRPF4;SELL;RGS1;SUB1;ALOX5;CCL5;S100A8 | |
| TLR2_ko Down | 25/245 | 3 × 10−8 | 3 × 10−7 | 4.04 | 69.7 | SPARC;FHL1;HTR2B;HBB;HNMT;FBLN5;MS4A4A;AKAP12;HHEX;PDGFD;ARHGDIB;PDK4;PDE8B;MSR1;ZNF160;SORBS1;PTK2;ACTA1;CXCL12;OAS1;NFIB;MPPED2;NOX4;PLIN2;CRK | |
| TLR3_ko Down | 13/243 | 2 × 10−2 | 2 × 10−2 | 1.97 | 7.6 | CD74;CD300A;CFI;HBB;HSPA2;TMEM135;DHRS9;SELL;OAS1;NAMPT;CASP1;PLIN2;S100A8 | |
| TLR4_ko Down | 17/243 | 6 × 10−4 | 1 × 10−3 | 2.64 | 19.7 | PHTF2;JUND;TACSTD2;CXCR4;HSPA2;NXT2;REXO2;ETS1;ZFP36L2;GULP1;PLAC8;RCAN1;MAPKAPK2;ANGPTL2;YPEL5;PDLIM5;S100A8 | |
| TLR5_ko Down | 16/243 | 2 × 10−3 | 2 × 10−3 | 2.47 | 15.9 | RERE;COL15A1;SPARC;NLGN4X;TRRAP;IGFBP2;TACSTD2;G0S2;DPT;TFPI;GULP1;AKAP12;SFRP4;PRPF4;ALOX5;PIEZO1 | |
| TLR5_ko Up | 19/247 | 8 × 10−5 | 2 × 10−4 | 2.93 | 27.6 | CD52;RAB1A;MEF2C;HPGD;DUSP1;USO1;YIPF5;TNFRSF11B;HK2;RHOB;ACTA1;CARHSP1;MYO1B;LOX;TNNT1;PXDN;ARHGDIB;PHACTR2;SNX5 | |
| TLR6_ko Down | 13/245 | 2 × 10−2 | 3 × 10−2 | 1.95 | 7.4 | C1S;RRAD;STAT1;TACSTD2;FHL2;CYBA;GLRX;HSPA2;PTK2;RCAN1;ADAMTS1;NAMPT;GLUL | |
| TLR6_ko Up | 12/245 | 5 × 10−2 | 5 × 10−2 | 1.79 | 5.5 | LAPTM4B;POSTN;MAF;SEMA3C;LMO3;NFATC3;BGN;HMGB1;ETS1;CHGB;PAFAH1B1;TBC1D16 | |
| TLR7_ko Down | 23/243 | 5 × 10−7 | 2 × 10−6 | 3.71 | 54.1 | CFD;COL15A1;C1S;PDE1A;FHL1;RNASE6;HNMT;TFPI;CP;MS4A4A;GULP1;PLAC8;MS4A6A;MFAP4;MAF;CXCL12;MDFIC;PDGFD;TXNIP;COL21A1;TCF4;HLA-DQA1;FGFR1 | |
| TLR7_ko Up | 15/242 | 4 × 10−3 | 5 × 10−3 | 2.31 | 12.8 | HSPA9;IRX5;BGN;TACSTD2;HK2;RHOB;PRPF4;ALOX5;QPCT;NUP50;ID1;ARHGDIB;GNB1;ITGA8;PIEZO1 | |
| TLR8_ko Down | 20/246 | 2 × 10−5 | 6 × 10−5 | 3.12 | 33.1 | CFD;DUSP4;BRD2;DDX3X;NLGN4X;MTCL1;ZNF160;RNASET2;FHL1;TACSTD2;CYBA;ZFP36L2;TBX3;AKAP12;MARCO;SNX1;NFIB;UST;GLUL;TGM2 | |
| TLR8_ko Up | 18/247 | 2 × 10−4 | 4 × 10−4 | 2.76 | 22.9 | PSMD10;CD52;CD74;SPON1;POSTN;CISH;LST1;YIPF5;RNASE6;CYBRD1;CRIP2;HNMT;PLAC8;LAPTM4B;MDFIC;LOX;TNNT1;DNASE1L3 | |
| TLR9_ko Down | 30/247 | 2 × 10−11 | 7 × 10−10 | 4.97 | 122.6 | CDS1;SPON1;HPGD;SEMA3C;MTCL1;TACSTD2;HNMT;ALOX5;PEA15;TIMM17A;PHACTR2;CRYM;TGM2;YWHAH;CPA3;CD52;BNIP3;HSPA2;SGPP1;RHOB;VEGFA;PTP4A1;MS4A6A;TYROBP;MDFIC;SUB1;QPCT;CCNG2;ID1;CNOT8 | |
| TLR9_ko Up | 22/246 | 2 × 10−6 | 8 × 10−6 | 3 × 100 | 45.5 | CFD;BRD2;PDGFRA;MEF2C;COL15A1;ZNF160;PDE1A;FHL1;CYBA;BMP6;HK2;PLAC8;AKAP12;SFRP4;RELN;MAF;CXCL12;PDE8B;ITM2A;KCNJ2;CHGB;FGFR1 | |
| TLR10_ko Down | 25/244 | 3 × 10−8 | 3 × 10−7 | 4 × 100 | 70.3 | GLRX;AEBP1;CAMKK2;PHACTR2;SKP2;CRYM;GLUL;LYN;CPA3;BRD2;CD52;AKIRIN1;ANGPT1;DUSP1;ZNF160;IGFBP2;CYBA;CP;SFRP4;RAB14;CCNG2;TXNIP;CTNNB1;TCF4;S100A8 | |
| TLR10_ko Up | 14/246 | 1 × 10−2 | 1 × 10−2 | 2.1 | 9.6 | SRGN;CD74;ENTPD1;GYPC;NAB1;TACSTD2;CYBRD1;AKAP12;LAPTM4B;HHEX;CXCL12;SELL;NFIB;RGS1 | |
| Fibrillation Down | TLR1_ko Up | 11/245 | 9 × 10−4 | 1 × 10−2 | 3.32 | 23.4 | MYBPC1;C7;PTPRZ1;CHL1;ANXA3;ANPEP;GREB1;SCNN1A;CHI3L1;SLC4A4;SORL1 |
| TLR4_ko Up | 9/246 | 9 × 10−3 | 4 × 10−2 | 2.66 | 12.4 | GRAMD1C;C7;ANXA3;SCNN1A;NR2F1;SLC4A4;SORL1;EREG;DCBLD2 | |
| TLR5_ko Down | 12/243 | 2 × 10−4 | 6 × 10−3 | 3.68 | 31.1 | SELENBP1;CADPS2;EHF;COL3A1;CHL1;ANPEP;GREB1;NR2F1;NINL;SORL1;SF1;CLGN | |
| TLR7_ko Down | 9/243 | 9 × 10−3 | 4 × 10−2 | 2.7 | 12.8 | AKR1B10;PPP1R1A;CALD1;ANPEP;RUFY3;P2RY14;RUFY2;STC1;AREG | |
| TLR7_ko Up | 9/242 | 8 × 10−3 | 4 × 10−2 | 2.71 | 12.9 | ERBB3;PTPRZ1;ANXA3;CHI3L1;SLC7A11;TUG1;CLDN1;SORL1;MFAP3L | |
| TLR8_ko Down | 10/246 | 3 × 10−3 | 2 × 10−2 | 2.98 | 17.3 | TKTL1;MYBPC1;SEMA6A;FLRT3;ANXA3;ACSL6;KRT7;AQP4;ZNF721;DSC3 | |
| TLR10_ko Up | 10/246 | 3 × 10−3 | 2 × 10−2 | 2.98 | 17.3 | TKTL1;BCHE;ARNT2;PTPRZ1;CHL1;ANXA3;KRT7;SLC4A4;EREG;FOSL2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagone, P.; Mangano, K.; Basile, M.S.; Munoz-Valle, J.F.; Perciavalle, V.; Nicoletti, F.; Bendtzen, K. Evaluation of Toll-like Receptor 4 (TLR4) Involvement in Human Atrial Fibrillation: A Computational Study. Genes 2024, 15, 634. https://doi.org/10.3390/genes15050634
Fagone P, Mangano K, Basile MS, Munoz-Valle JF, Perciavalle V, Nicoletti F, Bendtzen K. Evaluation of Toll-like Receptor 4 (TLR4) Involvement in Human Atrial Fibrillation: A Computational Study. Genes. 2024; 15(5):634. https://doi.org/10.3390/genes15050634
Chicago/Turabian StyleFagone, Paolo, Katia Mangano, Maria Sofia Basile, José Francisco Munoz-Valle, Vincenzo Perciavalle, Ferdinando Nicoletti, and Klaus Bendtzen. 2024. "Evaluation of Toll-like Receptor 4 (TLR4) Involvement in Human Atrial Fibrillation: A Computational Study" Genes 15, no. 5: 634. https://doi.org/10.3390/genes15050634
APA StyleFagone, P., Mangano, K., Basile, M. S., Munoz-Valle, J. F., Perciavalle, V., Nicoletti, F., & Bendtzen, K. (2024). Evaluation of Toll-like Receptor 4 (TLR4) Involvement in Human Atrial Fibrillation: A Computational Study. Genes, 15(5), 634. https://doi.org/10.3390/genes15050634