

Healthy Live Births after the Transfer of Mosaic Embryos: Self-Correction or PGT-A Overestimation?


Highlights
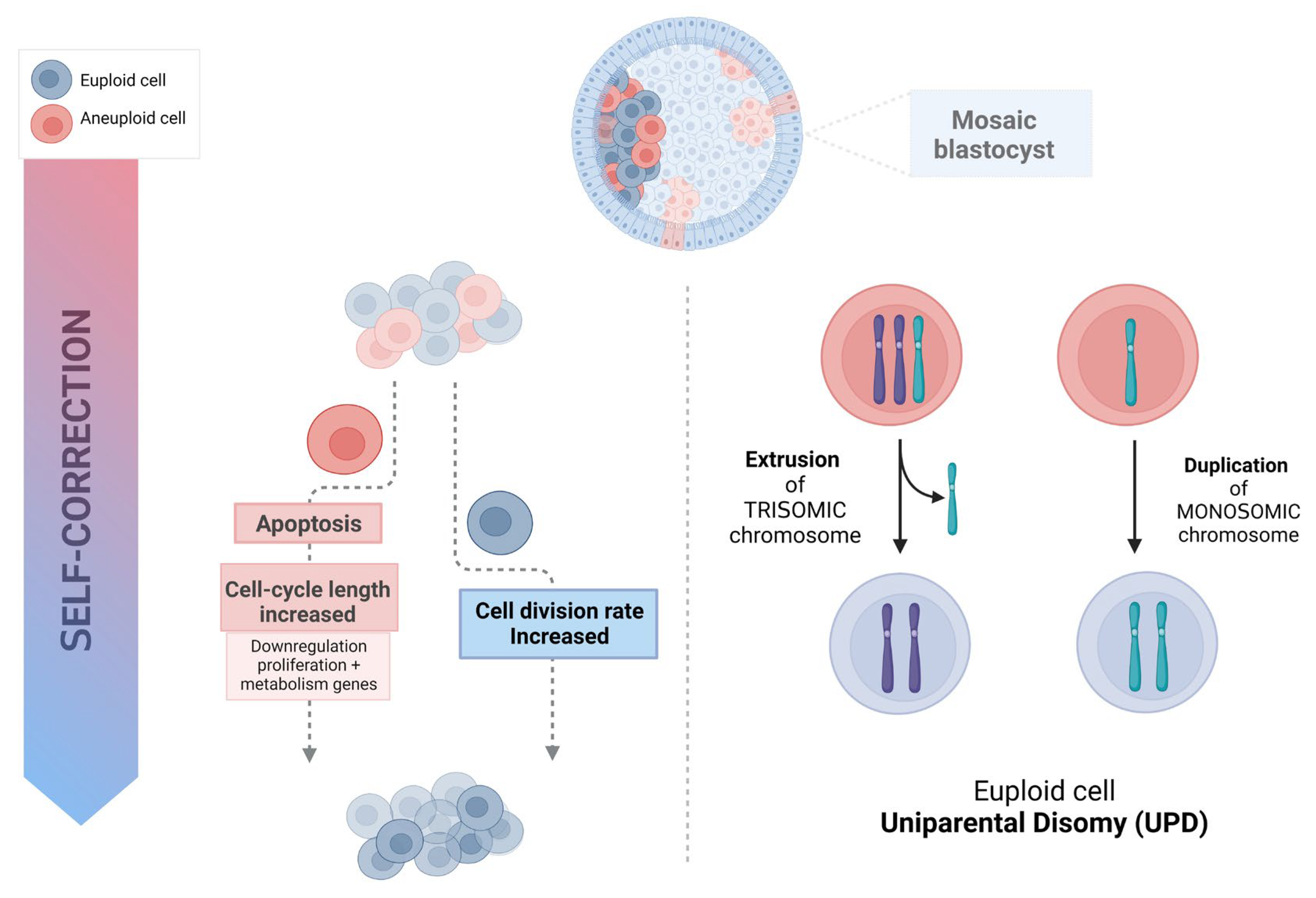
- A self-correction mechanism with preferential apoptosis in the ICM lineage in aneuploid-euploid mosaics, emerges as a potential mechanism to eliminate aneuploid cells during early embryo development.
- Assumptions of mosaicism from intermediate copy numbers in NGS-based PGT-A are inaccurate, identifying often as “mosaic,” embryos that in fact are uniformly euploid or aneuploid.
- Embryo plasticity represents a promising model of normalization to an euploid embryo constitution.
- It becomes mandatory to redesign the current technologies and explore novel methods to improve the accuracy of mosaicism diagnosis in PGT-A.
Abstract
:1. Introduction
2. Self-Correction and Embryo Plasticity
3. The Mortality Model (Apoptosis and Depletion)
4. Trisomy/Monosomy Rescue Model
5. Misinterpretations of PGT-A Results
5.1. Technical Accuracy
5.2. Concordance between TE and ICM
6. Discussion
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Sigurjonsson, S.; Pettersen, B.; Maisenbacher, M.K.; Hall, M.P.; Demko, Z.; Lathi, R.B.; Tao, R.; Aggarwal, V.; Rabinowitz, M. Genomic imbalance in products of conception: Single-nucleotide polymorphism chromosomal microarray analysis. Obstet. Gynecol. 2014, 124, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Wasielak-Politowska, M.; Kordowitzki, P. Chromosome Segregation in the Oocyte: What Goes Wrong during Aging. Int J Mol Sci. 2022, 23, 2880. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Mertzanidou, A.; Spits, C.; Nguyen, H.T.; Van De Velde, H.; Sermon, K. Evolution of aneuploidy up to Day 4 of human preimplantation development. Hum. Reprod. 2013, 28, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, A.W.; Tao, X.; Zhan, Y.; Whitehead, C.; Kim, J.; Hanson, B.; Osman, E.; Kim, T.J.; Patounakis, G.; Gutmann, J.; et al. A multicenter, prospective, blinded, nonselection study evaluating the predictive value of an aneuploid diagnosis using a targeted next-generation sequencing-based preimplantation genetic testing for aneuploidy assay and impact of biopsy. Fertil. Steril. 2021, 115, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Poli, M.; Rienzi, L.; Girardi, L.; Patassini, C.; Fabiani, M.; Cimadomo, D.; Benini, F.; Farcomeni, A.; Cuzzi, J.; et al. Mosaic human preimplantation embryos and their developmental potential in a prospective, non-selection clinical trial. Am. J. Hum. Genet. 2021, 108, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Munne, S.; Grifo, J.; Wells, D. Mosaicism: “Survival of the fittest” versus “no embryo left behind”. Fertil. Steril. 2016, 105, 1146–1149. [Google Scholar] [CrossRef]
- Rodrigo, L.; Clemente-Ciscar, M.; Campos Galindo, I.; Peinado, V.; Simon, C.; Rubio, C. Characteristics of the IVF Cycle that Contribute to the Incidence of Mosaicism. Genes 2020, 11, 1151. [Google Scholar] [CrossRef]
- Marin, D.; Xu, J.; Treff, N.R. Preimplantation genetic testing for aneuploidy: A review of published blastocyst reanalysis concordance data. Prenat. Diagn. 2021, 41, 545–553. [Google Scholar] [CrossRef]
- Huang, Y.; Ha, S.; Li, Z.; Li, J.; Xiao, W. CHKCENP B/MAD2 is associated with mild oxidative damage-induced sex chromosome aneuploidy of male mouse embryos during in vitro fertilization. Free Radic. Biol. Med. 2019, 137, 181–193. [Google Scholar] [CrossRef]
- Li, J.; Ha, S.; Li, Z.; Huang, Y.; Lin, E.; Xiao, W. Aurora B prevents aneuploidy via MAD2 during the first mitotic cleavage in oxidatively damaged embryos. Cell Prolif. 2019, 52, e12657. [Google Scholar] [CrossRef] [PubMed]
- Babariya, D.; Fragouli, E.; Alfarawati, S.; Spath, K.; Wells, D. The incidence and origin of segmental aneuploidy in human oocytes and preimplantation embryos. Hum. Reprod. 2017, 32, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.; Van de Velde, H.; De Paepe, C.; Sermon, K.; Spits, C. Mitotic spindle disruption in human preimplantation embryos activates the spindle assembly checkpoint but not apoptosis until day 5 of development. Mol. Hum. Reprod. 2017, 23, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Diez, C.; Paim, L.M.G.; Fitz Harris, G. Cell-sizeindependent spindle checkpoint failure underlies chromosome segregation error in mouse embryos. Curr. Biol. 2019, 29, 865.e3–873.e3. [Google Scholar] [CrossRef] [PubMed]
- Mantikou, E.; Wong, K.M.; Repping, S.; Mastenbroek, S. Molecular origin of mitotic aneuploidies in preimplantation embryos. Biochim. Et Biophys. Acta 2012, 1822, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Ye, Z.; Clarke, R.; Rosenwaks, Z.; Zaninovic, N. Direct unequal cleavages: Embryo developmental competence, genetic constitution and clinical outcome. PLoS ONE 2016, 11, e0166398. [Google Scholar] [CrossRef] [PubMed]
- Munne, S.; Blazek, J.; Large, M.; Martinez-Ortiz, P.A.; Nisson, H.; Liu, E.; Tarozzi, N.; Borini, A.; Becker, A.; Zhang, J.; et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of highresolution next-generation sequencing. Fertil. Steril. 2017, 108, 62–71. [Google Scholar] [CrossRef]
- Lin, P.Y.; Lee, C.I.; Cheng, E.H.; Huang, C.C.; Lee, T.H.; Shih, H.H.; Pai, Y.P.; Chen, Y.C.; Lee, M.S. Clinical Outcomes of Single Mosaic Embryo Transfer: High-Level or Low-Level Mosaic Embryo, Does it Matter? J. Clin. Med. 2020, 9, 1695. [Google Scholar] [CrossRef]
- Viotti, M.; Greco, E.; Grifo, J.A.; Madjunkov, M.; Librach, C.; Cetinkaya, M.; Kahraman, S.; Yakovlev, P.; Kornilov, N.; Corti, L.; et al. Chromosomal, gestational, and neonatal outcomes of embryos classified as a mosaic by preimplantation genetic testing for aneuploidy. Fertil. Steril. 2023, 120, 957–966. [Google Scholar] [CrossRef]
- Greco, E.; Yakovlev, P.; Kornilov, N. Two clinical case reports of embryonic mosaicism identified with PGT-A persisting during pregnancy as true fetal mosaicism. Hum. Reprod. 2023, 38, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Viotti, M.; McCoy, R.C.; Griffin, D.K.; Spinella, F.; Greco, E.; Madjunkov, M.; Madjunkova, S.; Librach, C.L.; Victor, A.R.; Barnes, F.L.; et al. Let the data do the talking: The need to consider mosaicism during embryo selection. Fertil. Steril. 2021, 116, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Marin, D. The “mosaic” embryo: Misconceptions and misinterpretations in preimplantation genetic testing for aneuploidy. Fertil. Steril. 2021, 116, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Minasi, M.G.; Fiorentino, F. Healthy Babies after Intrauterine Transfer of Mosaic Aneuploid Blastocysts. N. Engl. J. Med. 2015, 373, 2089–2090. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, S.; Cetinkaya, M.; Yuksel, B.; Yesil, M.; Pirkevi Cetinkaya, C. The birth of a baby with mosaicism resulting from a known mosaic embryo transfer: A case report. Hum. Reprod. 2020, 35, 727–733. [Google Scholar] [CrossRef]
- Schlade-Bartusia, K.; Strong, E.; Zhu, O.; Mackie, J.; Salema, D.; Volodarsky, M.; Roberts, J.; Steinraths, M. Mosaic embryo transfer—First report of a live born with non-mosaic partial aneuploidy and uniparental disomy 15. FS Rep. 2022, 3, 192–198. [Google Scholar]
- Van Echten-Arends, J.; Mastenbroek, S.; Sikkema-Raddatz, B.; Korevaar, J.C.; Heineman, M.J.; van der Veen, F.; Repping, S. Chromosomal mosaicism in human preimplantation embryos: A systematic review. Hum. Reprod. Update 2011, 17, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Malvestiti, F.; Agrati, C.; Grimi, B.; Pompilii, E.; Izzi, C.; Martinoni, L.; Gaetani, E.; Liuti, M.R.; Trotta, A.; Maggi, F.; et al. Interpreting mosaicism in chorionic villi: Results of a monocentric series of 1001 mosaics in chorionic villi with follow-up amniocentesis. Prenat. Diagn. 2015, 35, 1117–1127. [Google Scholar] [CrossRef]
- Barrie, A.; Homburg, R.; McDowell, G.; Brown, J.; Kingsland, C.; Troup, S. Preliminary investigation of the prevalence and implantation potential of abnormal embryonic phenotypes assessed using time-lapse imaging. Reprod. Biomed. Online 2017, 34, 455–462. [Google Scholar] [CrossRef]
- Lagalla, C.; Tarozzi, N.; Sciajno, R.; Wells, D.; Di, S.M.; Nadalini, M.; Distratis, V.; Borini, A. Embryos with morphokinetic abnormalities may develop into euploid blastocysts. Reprod. Biomed. Online 2017, 34, 137–146. [Google Scholar] [CrossRef]
- Coticchio, G.; Lagalla, C.; Sturmey, R.; Pennetta, F.; Borini, A. The enigmatic morula: Mechanisms of development, cell fate determination, self-correction and implications for ART. Hum. Reprod. Update 2019, 25, 422–438. [Google Scholar] [CrossRef]
- Coticchio, G.; Barrie, A.; Lagalla, C.; Borini, A.; Fishel, S.; Griffin, D.; Campbell, A. Plasticity of the human preimplantation embryo: Developmental dogmas, variations on themes and self-correction. Hum. Reprod. Update 2021, 27, 848–865. [Google Scholar] [CrossRef] [PubMed]
- Munne, S.; Velilla, E.; Colls, P.; Bermudez, M.G.; Vemuri, M.C.; Steuerwald, N.; Garrisi, J.; Cohen, J. Self Correction of chromosomally abnormal embryos in culture and implications or stem cell production. Fertil. Steril. 2005, 845, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Barbash-Hazan, S.; Frumkin, T.; Malcov, M.; Yaron, Y.; Cohen, T.; Azem, F.; Amit, A.; Ben-Yosef, D. Preimplantation aneuploid embryos undergo self-correction in correlation with their developmental potential. Fertil. Steril. 2009, 92, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M.; Dhaenens, L.; Taelman, J.; Dheedene, A.; Bialecka, M.; De Sutter, P.; Chuva de Sousa Lopes, S.M.; Menten, B.; Heindryckx, B. Extended in vitro culture of human embryos demonstrates the complex nature of diagnosing chromosomal mosaicism from a single trophectoderm biopsy. Hum. Reprod. 2019, 34, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Bolton, H.; Graham, S.J.L.; Van der Aa, N.; Kumar, P.; Theunis, K.; Gallardo, E.F.; Voet, T.; Zernicka-Goetz, M. Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploidy cells and normal developmental potential. Nat. Commun. 2016, 7, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Tighe, A.; D’Alise, A.M.; Taylor, S.S.; Musacchio, A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 2010, 190, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Rossant, J.; Tam, P.L. New insights into early human development: Lessons for stem cell derivation and differentiation. Cell Stem Cell 2017, 20, 18–28. [Google Scholar] [CrossRef]
- Yang, M.; Rito, T.; Metzger, J.; Naftaly, J.; Soman, R.; Hu, J.; Albertini, D.F.; Barad, D.H.; Brivanlou, A.H.; Gleicher, N. Depletion of aneuploid cells in human embryos and gastruloids. Nat. Cell Biol. 2021, 23, 314–321. [Google Scholar] [CrossRef]
- Victor, A.R.; Griffin, D.K.; Brake, A.J.; Tyndall, J.C.; Murphy, A.E.; Lepkowsky, L.T.; Lal, A.; Zouves, C.G.; Barnes, F.L.; McCoy, R.C.; et al. Assessment of aneuploidy concordance between clinical trophectoderm biopsy and blastocyst. Hum. Reprod. 2019, 34, 181–192. [Google Scholar] [CrossRef]
- Starostik, M.R.; Sosina, O.A.; McCoy, R.C. Single-cell analysis of human embryos reveals diverse patterns of aneuploidy and mosaicism. Genome Res. 2020, 30, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Roudebush, W.E.; Chosed, R.J. Embryo Biopsy can offer more information than just ploidy status. Front. Cell Dev. Biol. 2020, 8, 78. [Google Scholar] [CrossRef]
- Palini, S.; Galluzzi, L.; De Stefani, S.; Bianchi, M.; Wells, D.; Magnani, M.; Bulletti, C. Genomic DNA in human blastocoele fluid. Reprod. Biomed. Online 2013, 26, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Romanelli, V.; Patassini, C.; Poli, M.; Girardi, L.; Giancani, A.; Stoppa, M.; Cimadomo, D.; Ubaldi, F.M.; Rienzi, L. Diagnostic efficacy of blastocoel fluid and spent media as sources of DNA for preimplantation genetic testing in standard clinical conditions. Fertil. Steril. 2018, 110, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Tobler, K.J.; Zhao, Y.; Ross, R.; Benner, A.T.; Xu, X.; Du, L.; Broman, K.; Thrift, K.; Brezina, P.R.; Kearns, W.G. Blastocoel fluid from differentiated blastocysts harbors embryonic genomic material capable of a whole-genome deoxyribonucleic acid amplification and comprehensive chromosome microarray analysis. Fertil. Steril. 2015, 104, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.K.; Brezina, P.R.; Tobler, K.; Zhao, Y.; Silvestri, G.; Mccoy, R.C.; Anchan, R.; Benner, A.; Cutting, G.R.; Kearns, W.G. The human embryonic genome is karyotypically complex, with chromosomally abnormal cells preferentially located away from the developing fetus. Hum. Reprod. 2023, 38, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Athavale, D.M.; Barré, A.; Kranyak, A.C.; Lal, A.; Blalock, J.L.; Zimmerman, S.; Chang, T.A.; Robinson, R.D.; Wininger, J.D.; Roudebush, W.E.; et al. Pro-apoptotic gene expression in blastocoel fluid from euploid day-5 embryos is associated with negative pregnancy outcomes. Fertil. Steril. 2019, 112, e261. [Google Scholar] [CrossRef]
- Kranyak, A.C.; Barré, A.; Athavale, D.M.; Lal, A.; Blalock, J.L.; Zimmerman, S.; Chang, T.A.; Robinson, R.D.; Wininger, J.D.; Roudebush, W.E.; et al. Are there any similarities in gene expression between euploid embryos and aneuploid embryos compatible with life? Fertil. Steril. 2019, 112, e259. [Google Scholar] [CrossRef]
- Battaglia, R.; Palini, S.; Vento, M.E.; La Ferlita, A.; Lo Faro, M.J.; Caroppo, E.; Borzì, P.; Falzone, L.; Barbagallo, D.; Ragusa, M.; et al. Identification of extracellular vesicles and characterization of miRNAexpression profiles in human blastocoel fluid. Sci. Rep. 2019, 9, 84. [Google Scholar] [CrossRef]
- Gueye, N.A.; Devkota, B.; Taylor, D.; Pfundt, R.; Scott, R.T., Jr.; Treff, N.R. Uniparental disomy in the human blastocyst is exceedingly rare. Fertil. Steril. 2014, 101, 232–236. [Google Scholar] [CrossRef]
- Northrop, L.E.; Treff, N.R.; Levy, B.; Scott, R.T., Jr. SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocysts. Mol. Hum. Reprod. 2010, 16, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Nakka, P.; Smith, S.P.; O’Donnell-Luria, A.; McManus, K.F. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 2019, 105, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Scuffins, J.; Keller-Ramey, J.; Dyer, L.; Douglas, G.; Torene, R.; Gainullin, V.; Juusola, J.; Meck, J.; Retterer, K. Uniparental disomy in a population of 32,067 clinical exome trios. Genet. Med. 2021, 23, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Girardi, L.; Figliuzzi, M.; Poli, M.; Serdarogullari, M.; Patassini, C.; Caroselli, S.; Pergher, I.; Cogo, F.; Coban, O.; Boynukalin, F.K.; et al. The use of copy number loads to designate mosaicism in blastocyst stage PGT-A cycles: Fewer is better. Hum. Reprod. 2023, 16, dead049. [Google Scholar] [CrossRef] [PubMed]
- Victor, A.R.; Tyndall, J.C.; Brake, A.J.; Lepkowsky, L.T.; Murphy, A.E.; Griffin, D.K.; McCoy, R.C.; Barnes, F.L.; Zouves, C.G.; Viotti, M. One hundred mosaic embryos transferred prospectively in a single clinic: Exploring when and why they result in healthy pregnancies. Fertil. Steril. 2019, 111, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Palmerola, K.L.; Vitez, S.F.; Amrane, S.; Fischer, C.P.; Forman, E.J. Minimizing mosaicism: Assessing the impact of fertilization method on rate of mosaicism after next-generation sequencing (NGS) preimplantation genetic testing for aneuploidy (PGT-A). J. Assist. Reprod. Genet. 2019, 36, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Handyside, A.H.; McCollin, A.; Summers, M.C.; Ottolini, C.S. Copy number analysis of meiotic and postzygotic mitotic aneuploidies in trophectoderm cells biopsied at the blastocyst stage and arrested embryos. Prenat. Diagn. 2021, 41, 525–535. [Google Scholar] [CrossRef]
- Xiong, S.; Liu, W.; Wang, J.; Liu, J.; Gao, Y.; Wu, L.; Zhu, J.; Hao, X.; Li, J.; Liu, D.; et al. Trophectodrm biopsy protocolsmay impact the rate of mosaic blastocysts in cycles with preimplantation genetic testing foraneuploidy. J. Assist. Reprod. Genet. 2021, 38, 1153–1162. [Google Scholar] [CrossRef]
- García-Pascual, C.M.; Navarro-Sánchez, L.; Navarro, R.; Martínez, L.; Jiménez, J.; Simón, C.; Rubio, C. Optimized NGS Approach for Detection of Aneuploidies and Mosaicism in PGT-A and Imbalances in PGT-SR. Genes 2020, 11, 724. [Google Scholar] [CrossRef]
- Goodrich, D.; Xing, T.; Tao, X.; Lonczak, A.; Zhan, Y.; Landis, J.; Zimmerman, R.; Scott, R.T., Jr.; Treff, N.R. Evaluation of comprehensive chromosome screening platforms for the detection of mosaic segmental aneuploidy. J. Assist. Reprod. Genet. 2017, 34, 975–981. [Google Scholar] [CrossRef]
- Gleicher, N.; Orvieto, R. Is the hypothesis of preimplantation genetic screening (PGS) still supportable? A review. J. Ovarian Res. 2017, 10, 21. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Sun, X.; Liu, L.; Jin, H.; Li, M.; Witz, C.; Williams, D.; Griffith, J.; Skorupski, J.; et al. DNA microarray reveals that high proportions of human blastocysts from women of advanced maternal age are aneuploid and mosaic. Biol. Reprod. 2012, 87, 148. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jin, L.; Chen, W.; Liu, J.M.; Hu, J.; Yu, Q.; Ren, X.L.; Huang, B.; He, H. The true incidence of chromosomal mosaicism after preimplantation genetic testing is much lower than that indicated by trophectoderm biopsy. Hum. Reprod. 2021, 36, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Lenzi, M.; Ross, R.; Katz-Jaffe, M.; Schoolcraft, W.B.; Wells, D. Comprehensive molecular cytogenetic analysis of the human blastocyst stage. Hum. Reprod. 2008, 23, 2596–2608. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Wright, G.; Elliott, T.; Ubaldi, F.M.; Rienzi, L.; Nagy, Z.P. FISH reanalysis of inner cell mass and trophectoderm samples of previously array-CGH screened blastocysts shows high accuracy of diagnosis and no major diagnostic impact of mosaicism at the blastocyst stage. Hum. Reprod. 2013, 28, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yan, L.; Lu, S.; Zhao, N.; Qiao, J. Re-analysis of aneuploidy blastocysts with an inner cell mass and different regional trophectoderm cells. J. Assist. Reprod. Genet. 2017, 34, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Rienzi, L. Mosaicism between trophectoderm and inner cell mass. Fertil. Steril. 2017, 107, 1098–1106. [Google Scholar] [CrossRef]
- Kim, J.; Tao, X.; Cheng, M.; Steward, A.; Guo, V.; Zhan, Y.; Scott, R.T., Jr.; Jalas, C. The concordance rates of an initial trophectoderm biopsy with the rest of the embryo using PGTseq, a targeted next-generation sequencing platform for preimplantation genetic testing-aneuploidy. Fertil. Steril. 2022, 117, 315–323. [Google Scholar] [CrossRef]
- Tšuiko, O.; Zhigalina, D.I.; Jatsenko, T.; Skryabin, N.A.; Kanbekova, O.R.; Artyukhova, V.G.; Svetlakov, A.V.; Teearu, K.; Trošin, A.; Salumets, A.; et al. Karyotype of the blastocoel fluid demonstrates low concordance with both trophectoderm and inner cell mass. Fertil. Steril. 2018, 109, 1127–1134.e1. [Google Scholar] [CrossRef]
- Chuang, T.H.; Hsieh, J.Y.; Lee, M.J.; Lai, H.H.; Hsieh, C.L.; Wang, H.L.; Chang, Y.J.; Chen, S.U. Concordance between different trophectoderm biopsy sites and the inner cell mass of chromosomal composition measured with a next-generation sequencing platform. Mol. Hum. Reprod. 2018, 24, 593–601. [Google Scholar] [CrossRef]
- Lawrenz, B.; El Khatib, I.; Liñán, A.; Bayram, A.; Arnanz, A.; Chopra, R.; De Munck, N.; Fatemi, H.M. The clinicians’ dilemma with mosaicism-an insight from inner cell mass biopsies. Hum. Reprod. 2019, 34, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, N.M.; McCulloh, D.H.; Kramer, Y.; Keefe, D.; Grifo, J.A. The reproducibility of trophectoderm biopsies in euploid, aneuploid, and mosaic embryos using independently verified next-generation sequencing (NGS): A pilot study. J. Assist. Reprod. Genet. 2020, 37, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chavli, E.; van den Born, M.; Eleveld, C.; Boter, M.; van Marion, R.; Hoefsloot, L.; Laven, J.; Baart, E.; Van Opstal, D. Chromosomal mosaicism in human blastocysts: A cytogenetic comparison of trophectoderm and inner cell mass after next-generation sequencing. Reprod. Biomed. Online 2022, 45, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Werner, M.D.; Franasiak, J.M.; Forman, E.J.; Upham, K.; Treff, N.R.; Scott, R. Prolonged time to first cytokinesis and the interval between the five cell stage and early cavitation are associated with embryonic mosaicism. Fertil. Steril. 2014, 102, e209. [Google Scholar] [CrossRef]
- Sciorio, R.; Meseguer, M. Focus on time-lapse analysis: Blastocyst collapse and morphometric assessment as new features of embryo viability. Reprod. Biomed. Online 2021, 43, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Zahn, H.; Steif, A.; Laks, E.; Eirew, P.; Van Insberghe, M.; Shah, S.P.; Aparicio, S.; Hansen, C.L. Scalable whole-genome single-cell library preparation without preamplification. Nat. Methods 2017, 14, 167–173. [Google Scholar] [CrossRef]

{kind=link}
| PGT-A | Cases | Correlation | |||
|---|---|---|---|---|---|
| Euploidy | Aneuploidy | Mosaicism | |||
| Fragouli et al., 2008 [64] | FISH-aCGH | 10 | 100% (4/4) | 100% (6/6) | - |
| Northrop et al., 2010 [51] | SNP array | 21 | 42.8% (9/21) | ||
| Capalbo et al., 2013 [65] | FISH aCGH | 85 | 100% (20/20) | 90.2% (46/51) | 85.7% (12/14) |
| Huang et al., 2017 [66] | aCGH | 51 | - | 84.3% (43/51) | - |
| Victor et al., 2019 [40] | NGS | 93 | - | 96.8% (90/93) | - |
| Liu et al., 2012 [62] | aCGH | 13 | - | 30.8% (4/13) | - |
| Tsuiko et al., 2018 [69] | NGS | 14 | 100% (9/9) | 100% (2/2) | 75.0% (3/4) |
| Popovic et al., 2019 [35] | NGS | 24 | - | 100% (8/8) | 18.8% (3/16) |
| Chuang et al., 2018 [70] | NGS | 29 | 100% (8/8) | 43.8% (7/16) | 20.0% (1/5) |
| Wu et al., 2021 [63] | NGS | 91 | - | - | 27.5% (25/91) |
| Victor et al., 2019 [55] | NGS | 8 | - | 100% (3/3) | 40.0% (2/5) |
| Euploid 60.0% (3/5) | |||||
| Lawrenz et al., 2019 [71] | NGS | 84 | 93.2% (41/44) | 92.5% (37/40) | - |
| Sachdev et al., 2020 [72] | NGS | 32 | 99.5% | 97.3% | 35.2% |
| Lin et al., 2020 [19] | NGS | 14 | 50.0% (7/14) | ||
| Low-range mosaic | Euploid 50.0% (7/14) | ||||
| Aneuploid 0% (0/24) | |||||
| High-range mosaic | NGS | 27 | 37.0 (10/27) | ||
| Euploid 40.7% (11/27) | |||||
| Aneuploid 22.0% (6/27) | |||||
| Capalbo et al., 2021 [7] | NGS | ||||
| Low-range mosaic (20–30%) | 37 | - | - | 0% (1/148) | |
| Euploid 100% (147/148) | |||||
| Medium-range mosaic (30–50%) | 31 | - | - | 4.4% (2/46) | |
| Euploid 93.4% (43/46) | |||||
| Aneuploid 2.2% (1/46) | |||||
| High-range mosaic (50–70%) | 5 | - | - | 20.0% (4/20) | |
| Euploid 15.0% (3/20) | |||||
| Aneuploid 65.0% (13/20) | |||||
| Chavli et al., 2022 [73] | NGS | ||||
| Low-range mosaic | 3 | 0% (0/3) | |||
| Euploid 100% (3/3) | |||||
| High-range mosaic | 4 | 25.0% (1/4) | |||
| Euploid 25.0% (1/4) | |||||
| Aneuploid 50.0% (2/4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, G.; Sciorio, R.; Fleming, S. Healthy Live Births after the Transfer of Mosaic Embryos: Self-Correction or PGT-A Overestimation? Genes 2024, 15, 18. https://doi.org/10.3390/genes15010018
Campos G, Sciorio R, Fleming S. Healthy Live Births after the Transfer of Mosaic Embryos: Self-Correction or PGT-A Overestimation? Genes. 2024; 15(1):18. https://doi.org/10.3390/genes15010018
Chicago/Turabian StyleCampos, Gerard, Romualdo Sciorio, and Steven Fleming. 2024. "Healthy Live Births after the Transfer of Mosaic Embryos: Self-Correction or PGT-A Overestimation?" Genes 15, no. 1: 18. https://doi.org/10.3390/genes15010018
APA StyleCampos, G., Sciorio, R., & Fleming, S. (2024). Healthy Live Births after the Transfer of Mosaic Embryos: Self-Correction or PGT-A Overestimation? Genes, 15(1), 18. https://doi.org/10.3390/genes15010018