
Genome-Wide Association Analysis Unravels New Quantitative Trait Loci (QTLs) for Eight Lodging Resistance Constituent Traits in Rice (Oryza sativa L.)




Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Field Experiments
2.2. Phenotypic Investigation for Eight Lodging Resistance Traits
2.2.1. Stem Anti-Thrust
2.2.2. Plant Height
2.2.3. Stem Diameter and Internode Length
2.2.4. SSR Marker Genotyping
2.2.5. Heritability
2.2.6. Genetic Phylogenetic and Population Structure Analysis
2.2.7. Linkage Disequilibrium Analysis
2.2.8. Association Mapping
3. Results
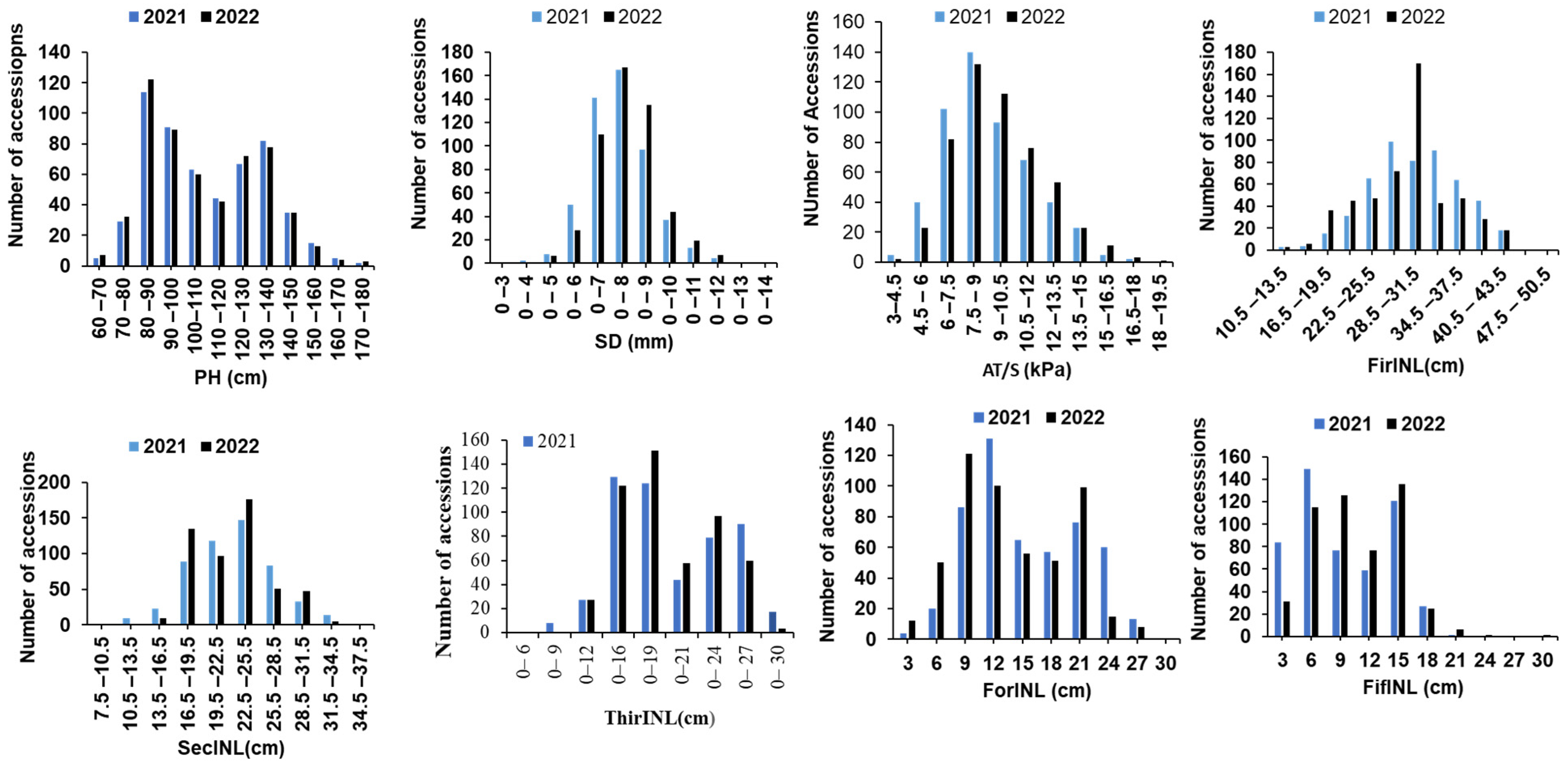
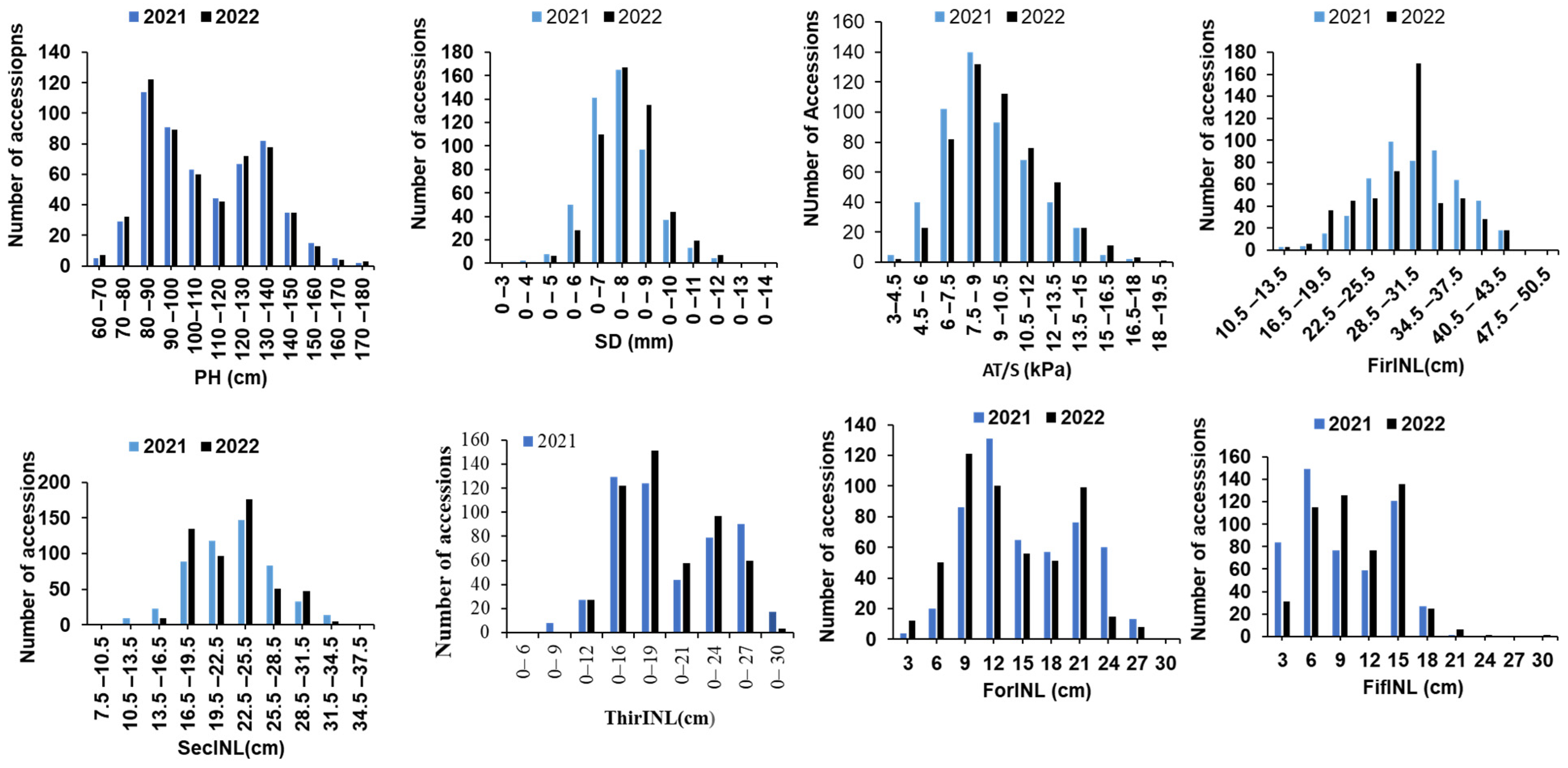
3.1. Phenotypic Evaluations
3.2. Genetic Diversity
3.3. Population Structure and Genetic Relatedness
3.4. Genetic Differentiation among Subpopulations
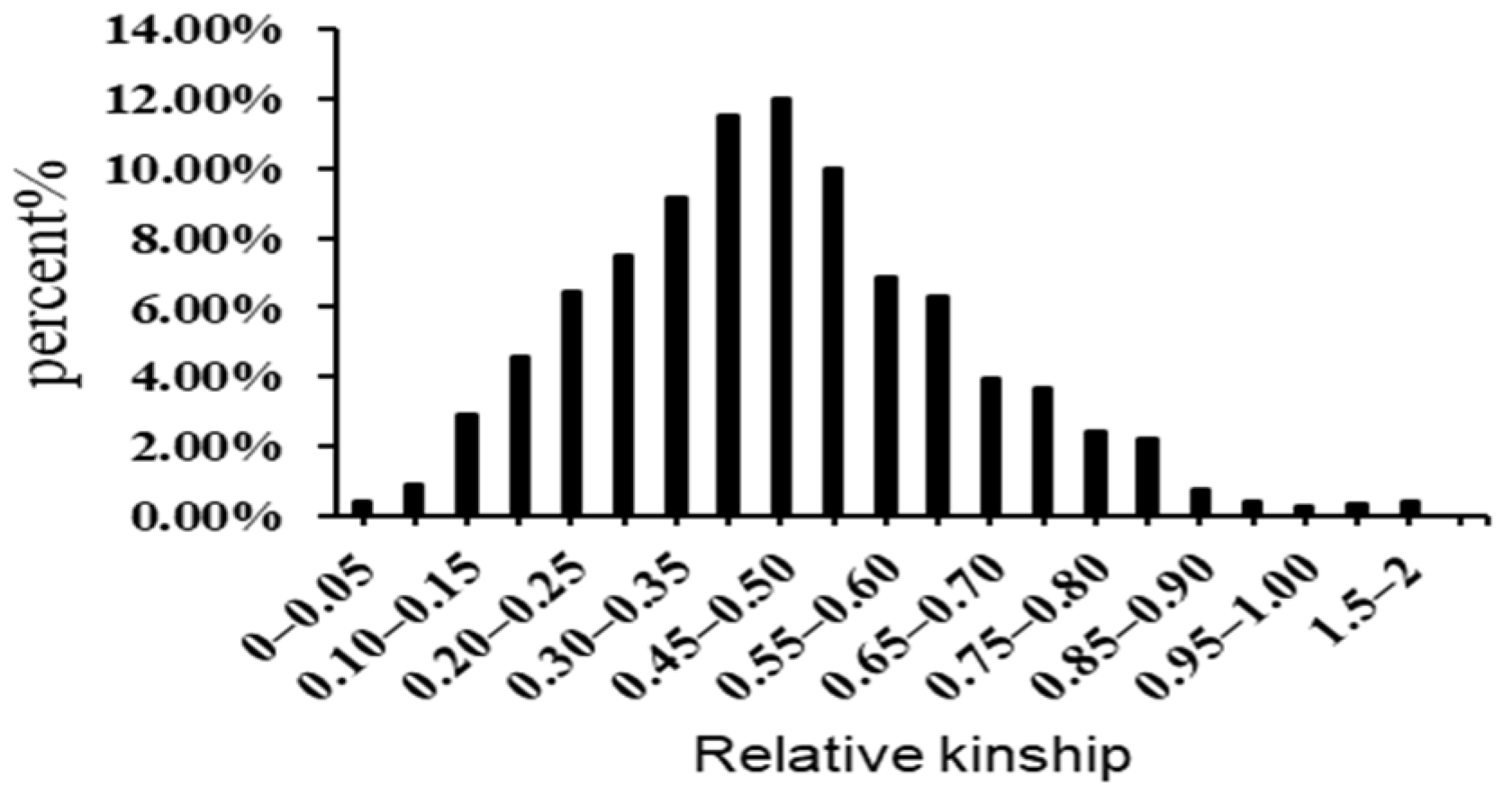
3.5. Linkage Disequilibrium
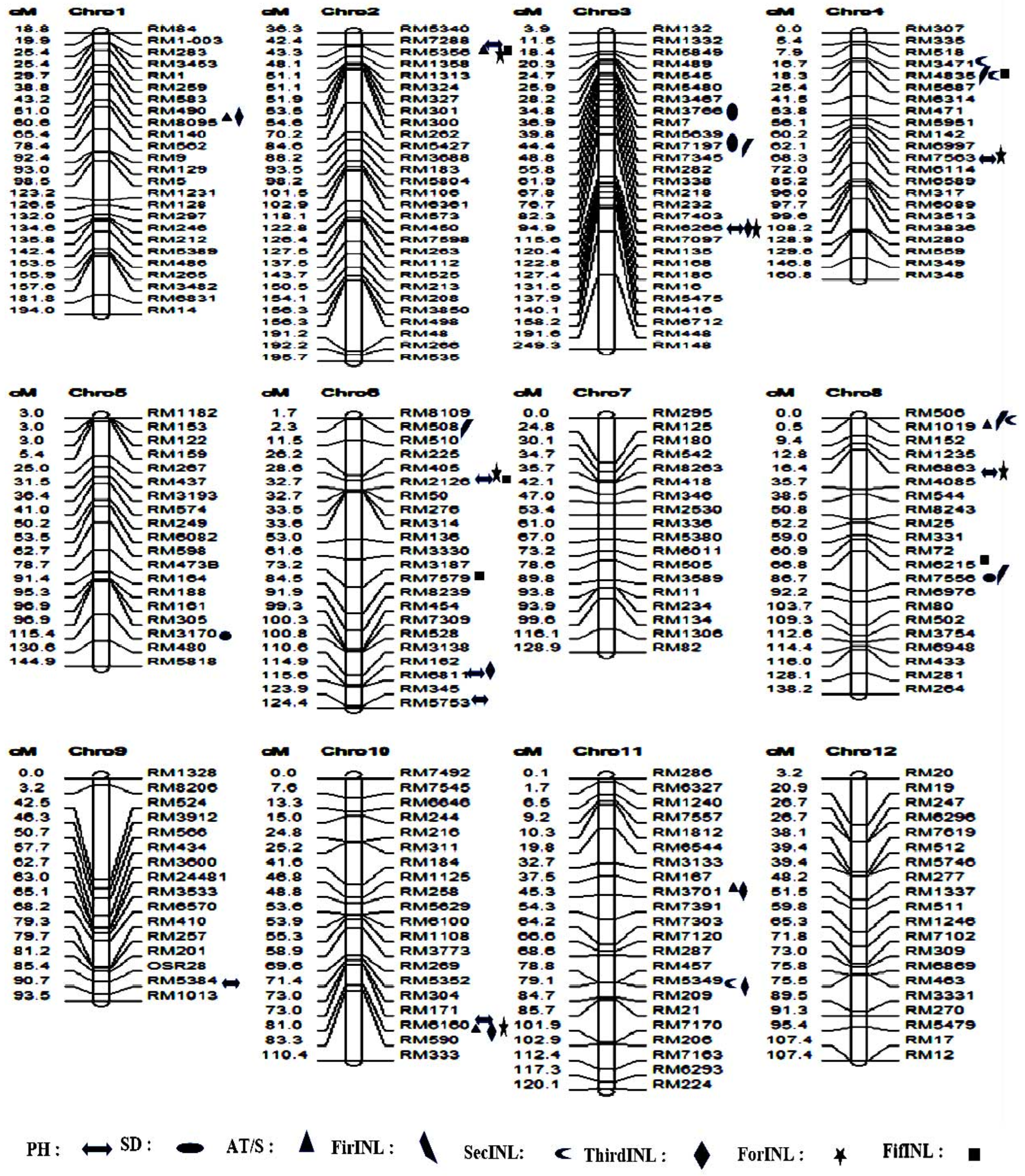
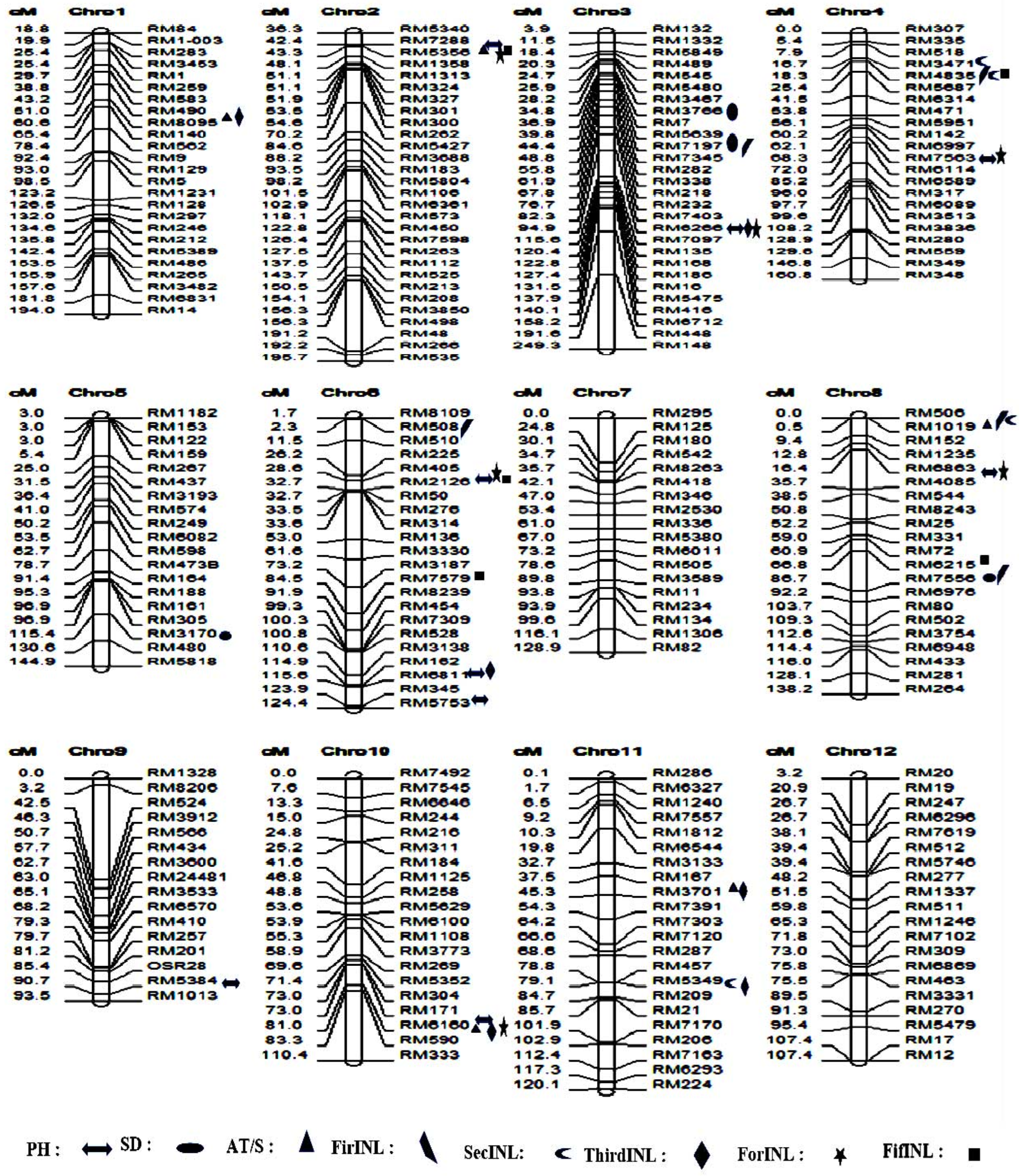
3.6. Discovery of Marker–Trait Associations and Favorable Alleles for the Eight Traits in a Natural Population
3.7. SSR Association Loci and Favorable Alleles for Various Plant Traits
3.7.1. Plant Height in the Natural Population
3.7.2. Stem Diameter in the Natural Population
3.7.3. Stem Anti-Thrust in the Natural Population
3.7.4. First Internode Length Trait (FirINL) in the Natural Population
3.7.5. Second Internode Length Trait (SecINL) in the Natural Population
3.7.6. Third Internode Length Trait (ThirINL) in the Natural Population
3.7.7. Fourth Internode Length (ForINL) in the Natural Population
3.7.8. Fifth Internode Length Trait (FifINL) in the Natural Population
3.8. New QTLs Detected for the 8 Traits
3.9. Parental Combinations Predicted for Lodging-Resistant Improvement
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peña, D.; Martín, C.; Fernández-Rodríguez, D.; Terrón-Sánchez, J.; Vicente, L.A.; Albarrán, Á.; Rato-Nunes, J.M.; López-Piñeiro, A. Medium-Term Effects of Sprinkler Irrigation Combined with a Single Compost Application on Water and Rice Productivity and Food Safety. Plants 2023, 12, 456. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, T.; Sasaki, H.; Ishimaru, K. Factors Responsible for Decreasing Sturdiness of the Lower Part in Lodging of Rice (Oryza sativa L.). Plant Prod. Sci. 2005, 8, 166–172. [Google Scholar] [CrossRef]
- Shah, L.; Yahya, M.; Shah, S.M.A.; Nadeem, M.; Ali, A.; Ali, A.; Wang, J.; Riaz, M.W.; Rehman, S.; Wu, W.; et al. Improving Lodging Resistance: Using Wheat and Rice as Classical Examples. Int. J. Mol. Sci. 2019, 20, 4211. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Chen, T.; Zhao, C.; Zhou, M. Lodging Prevention in Cereals: Morphological, Biochemical, Anatomical Traits and Their Molecular Mechanisms, Management and Breeding Strategies. Field Crop. Res. 2022, 289, 108733. [Google Scholar] [CrossRef]
- Gardiner, B.; Berry, P.; Moulia, B. Review: Wind Impacts on Plant Growth, Mechanics and Damage. Plant Sci. 2016, 245, 94–118. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Laza, M.R.C.; Mendez, K.V.; Bhosale, S.; Dingkuhn, M. The Blaster: A Methodology to Induce Rice Lodging at Plot Scale to Study Lodging Resistance. Field Crop. Res. 2020, 245, 107663. [Google Scholar] [CrossRef]
- Zsögön, A.; Peres, L.E.P.; Xiao, Y.; Yan, J.; Fernie, A.R. Enhancing Crop Diversity for Food Security in the Face of Climate Uncertainty. Plant J. 2022, 109, 402–414. [Google Scholar] [CrossRef]
- Li, L.; He, L.; Li, Y.; Wang, Y.; Ashraf, U.; Hamoud, Y.A.; Hu, X.; Wu, T.; Tang, X.; Pan, S. Deep Fertilization Combined with Straw Incorporation Improved Rice Lodging Resistance and Soil Properties of Paddy Fields. Eur. J. Agron. 2023, 142, 126659. [Google Scholar] [CrossRef]
- Banan, D. Phenotypic and Genetic Variation in the Architectural Responses of a C4 Grass to Drought Stress. Ph.D. Dissertation, University of Illinois at Urbana-Champaign, Urbana, IL, USA, 2019. [Google Scholar]
- Sowadan, O.; Li, D.; Zhang, Y.; Zhu, S.; Hu, X.; Bhanbhro, L.B.; Edzesi, W.M.; Dang, X.; Hong, D. Mining of Favorable Alleles for Lodging Resistance Traits in Rice (Oryza sativa) through Association Mapping. Planta 2018, 248, 155–169. [Google Scholar] [CrossRef]
- Cai, W.; Hong, J.; Liu, Z.; Wang, W.; Zhang, J.; An, G.; Liang, W.; Persson, S.; Zhang, D. A Receptor-like Kinase Controls the Amplitude of Secondary Cell Wall Synthesis in Rice. Curr. Biol. 2023, 33, 498–506.e6. [Google Scholar] [CrossRef]
- Huang, N.; Courtois, B.; Khush, G.S.; Lin, H.; Wang, G.; Wu, P.; Zheng, K. Association of Quantitative Trait Loci for Plant Height with Major Dwarfing Genes in Rice. Heredity 1996, 77, 130–137. [Google Scholar] [CrossRef]
- Cao, G.; Zhu, J.; He, C.; Gao, Y.; Yan, J.; Wu, P. Impact of Epistasis and QTL×environment Interaction on the Developmental Behavior of Plant Height in Rice (Oryza sativa L.). Theor. Appl. Genet. 2001, 103, 153–160. [Google Scholar] [CrossRef]
- Hittalmani, S.; Shashidhar, H.E.; Bagali, P.G.; Huang, N.; Sidhu, J.S.; Singh, V.P.; Khush, G.S. Molecular Mapping of Quantitative Trait Loci for Plant Growth, Yield and Yield Related Traits across Three Diverse Locations in a Doubled Haploid Rice Population. Euphytica 2002, 125, 207–214. [Google Scholar] [CrossRef]
- Luo, A.; Qian, Q.; Yin, H.; Liu, X.; Yin, C.; Lan, Y.; Tang, J.; Tang, Z.; Cao, S.; Wang, X.; et al. EUI1, Encoding a Putative Cytochrome P450 Monooxygenase, Regulates Internode Elongation by Modulating Gibberellin Responses in Rice. Plant Cell Physiol. 2006, 47, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.M.; Wu, Y.J.; Fu, X.D.; Qian, Q. Characterizations and Molecular Mapping of a Novel Dominant Semi-dwarf Gene Sdd(t) in Rice (Oryza sativa). Plant Breed. 2008, 127, 125–130. [Google Scholar] [CrossRef]
- Miura, K.; Wu, J.; Sunohara, H.; Wu, X.; Matsumoto, T.; Matsuoka, M.; Ashikari, M.; Kitano, H. High-Resolution Mapping Revealed a 1.3-Mbp Genomic Inversion in Ssi1, a Dominant Semidwarf Gene in Rice (Oryza sativa). Plant Breed. 2009, 128, 63–69. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of Polyploid Wheat Genomic Diversity Using a High-Density 90 000 Single Nucleotide Polymorphism Array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef]
- Wang, J.; Gang, S.; Yang, L.; Zheng, H.; Sun, J.; Liu, H.; Zhao, H.; Zou, D. Markers Associated with Culm Length and Elongated Internode Length in Japonica Rice. Crop. Sci. 2017, 57, 2329–2344. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Yu, Y.; Ji, H.; Lee, G.-S.; Hyung, N.-I.; Lee, K.; Kim, T.-H. Fine-Mapping of a Major Quantitative Trait Locus q2ID1 for Rice Stem Diameter. Plant Breed. Biotech. 2021, 9, 298–309. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Ishimaru, K. Identification and Functional Analysis of a Locus for Improvement of Lodging Resistance in Rice. Plant Physiol. 2004, 134, 676–683. [Google Scholar] [CrossRef]
- Desai, H.; Hamid, R.; Ghorbanzadeh, Z.; Bhut, N.; Padhiyar, S.M.; Kheni, J.; Tomar, R.S. Genic Microsatellite Marker Characterization and Development in Little Millet (Panicum sumatrense) Using Transcriptome Sequencing. Sci. Rep. 2021, 11, 20620. [Google Scholar] [CrossRef]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and Experimental Analysis of Microsatellites in Rice (Oryza sativa L.): Frequency, Length Variation, Transposon Associations, and Genetic Marker Potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R.; Teytelman, L.; Xu, Y.; Lobos, K.B.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.; Xing, Y.; et al. Development and Mapping of 2240 New SSR Markers for Rice (Oryza sativa L.). DNA Res. 2002, 9, 199–207. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic Microsatellite Markers in Plants: Features and Applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Li, Q.; Su, X.; Ma, H.; Du, K.; Yang, M.; Chen, B.; Fu, S.; Fu, T.; Xiang, C.; Zhao, Q.; et al. Development of Genic SSR Marker Resources from RNA-Seq Data in Camellia Japonica and Their Application in the Genus Camellia. Sci. Rep. 2021, 11, 9919. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid Isolation of High Molecular Weight Plant DNA. Nucleic. Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.-H.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A Toolbox for Integrative Analysis and Visualization of Spatial Expression Data. Genome Biol. 2021, 22, 78. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Zheng, Y.; Han, X.; Zhao, Y.; Zhu, L.; Huang, Y.; Jia, X.; Zhang, Z.; Chen, J.; Guo, J. Association Mapping for General Combining Ability with Yield, Plant Height and Ear Height Using F1 Population in Maize. PLoS ONE 2021, 16, e0258327. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An Integrated Analysis Environment for Genetic Marker Analysis. Bioinform 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Dominant Markers and Null Alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of Estimated Phylogenetic Trees from Molecular Data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, J.; Zhao, H.; Liu, H.; Sun, J.; Guo, L.; Zou, D. Genetic Structure, Linkage Disequilibrium and Association Mapping of Salt Tolerance in Japonica Rice Germplasm at the Seedling Stage. Mol. Breed. 2015, 35, 152. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (Version 3.0): An Integrated Software Package for Population Genetics Data Analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A Unified Mixed-Model Method for Association Mapping That Accounts for Multiple Levels of Relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Salas Fernandez, M.G.; Bao, Y.; Tang, L.; Schnable, P.S. A High-Throughput, Field-Based Phenotyping Technology for Tall Biomass Crops. Plant Physiol. 2017, 174, 2008–2022. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association Mapping of Kernel Size and Milling Quality in Wheat (Triticum aestivum L.) Cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef]
- Liu, C.; Song, J.; Wang, Y.; Huang, X.; Zhang, F.; Wang, W.; Xu, J.; Zhang, Y.; Yu, H.; Pang, Y.; et al. Rapid Prediction of Head Rice Yield and Grain Shape for Genome-Wide Association Study in Indica Rice. J. Cereal. Sci. 2020, 96, 103091. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Guo, J.; Jing, S.; Zhu, L.; He, G. Fine Mapping of the Rice Brown Planthopper Resistance Gene BPH7 and Characterization of Its Resistance in the 93-11 Background. Euphytica 2014, 198, 369–379. [Google Scholar] [CrossRef]
- Dang, X.; Fang, B.; Chen, X.; Li, D.; Sowadan, O.; Dong, Z.; Liu, E.; She, D.; Wu, G.; Liang, Y.; et al. Favorable Marker Alleles for Panicle Exsertion Length in Rice (Oryza sativa L.) Mined by Association Mapping and the RSTEP-LRT Method. Front. Plant Sci. 2017, 8, 2112. [Google Scholar] [CrossRef] [PubMed]
- Multani, D.S.; Jiao, S.; Jung, M.T.; Simcox, K.D. Stalk Strength Improvement in Crop Plants: A Progress Report. Annu. Plant Rev. Online 2021, 4, 357–396. [Google Scholar] [CrossRef]
- Kashiwagi, T. Novel QTL for Lodging Resistance, PRL4, Improves Physical Properties with High Non-Structural Carbohydrate Accumulation of Basal Culms in Rice (Oryza sativa L.). Euphytica 2022, 218, 83. [Google Scholar] [CrossRef]
- Wang, J.; Yang, W.; Zhang, S.; Hu, H.; Yuan, Y.; Dong, J.; Chen, L.; Ma, Y.; Yang, T.; Zhou, L.; et al. A Pangenome Analysis Pipeline Provides Insights into Functional Gene Identification in Rice. Genome Biol. 2023, 24, 19. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.G.; Carabalí, S.J.; Giraldo, O.X.; Martínez, C.P.; Correa, F.; Prado, G.; Tohme, J.; Lorieux, M. Identification of a Rice stripe necrosis Virus Resistance Locus and Yield Component QTLs Using Oryza sativa × O. glaberrima Introgression Lines. BMC Plant Biol. 2010, 10, 6. [Google Scholar] [CrossRef]
- Li, J.; Xiao, J.; Grandillo, S.; Jiang, L.; Wan, Y.; Deng, Q.; Yuan, L.; McCouch, S.R. QTL Detection for Rice Grain Quality Traits Using an Interspecific Backcross Population Derived from Cultivated Asian (O. sativa L.) and African (O. glaberrima S.) Rice. Genome 2004, 47, 697–704. [Google Scholar] [CrossRef]
- Ishimaru, K.; Ono, K.; Kashiwagi, T. Identification of a New Gene Controlling Plant Height in Rice Using the Candidate-Gene Strategy. Planta 2004, 218, 388–395. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Togawa, E.; Hirotsu, N.; Ishimaru, K. Improvement of Lodging Resistance with QTLs for Stem Diameter in Rice (Oryza sativa L.). Theor. Appl. Genet. 2008, 117, 749–757. [Google Scholar] [CrossRef]
- Madoka, Y.; Kashiwagi, T.; Hirotsu, N.; Ishimaru, K. Indian Rice “Kasalath” Contains Genes That Improve Traits of Japanese Premium Rice “Koshihikari”. Theor. Appl. Genet. 2008, 116, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Singh, U.M.; Naik, S.M.; Venkateshwarlu, C.; Ramayya, P.J.; Raman, K.A.; Sandhu, N.; Kumar, A. Molecular Mapping of QTLs Associated with Lodging Resistance in Dry Direct-Seeded Rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 01431. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Jeong, I.-S.; Ji, H.; Lee, G.-S.; Yoon, U.-H.; Kim, T.-H. Development of New CAPS Markers and Their Application in QTL Analysis of Stem Diameter in Rice. Korean J. Breed. Sci. 2014, 46, 116–128. [Google Scholar] [CrossRef]
- Zhu, L.H.; Zhong, D.B.; Xu, J.L.; Yu, S.B.; Li, Z.K. Differential Expression of Lodging Resistance Related QTLs in Rice (Oryza sativa L.). Plant Sci. 2008, 175, 898–905. [Google Scholar] [CrossRef]
- Zha, R.; Yang, Z.; Zhao, F.; Sang, X.; Ling, Y.; Xie, R.; He, G. Prediction of F1 yield using genetic effects of molecular marker in indica rice (Oryza sativa L.). J. Plant Genet. Resour. 2010, 11, 72–77. [Google Scholar] [CrossRef]
- Jiang, C.-J.; Shimono, M.; Maeda, S.; Inoue, H.; Mori, M.; Hasegawa, M.; Sugano, S.; Takatsuji, H. Suppression of the Rice Fatty-Acid Desaturase Gene OsSSI2 Enhances Resistance to Blast and Leaf Blight Diseases in Rice. Mol. Plant Microbe Interact. 2009, 22, 820–829. [Google Scholar] [CrossRef]
- Yamamoto, T.; Taguchi-Shiobara, F.; Ukai, Y.; Sasaki, T.; Yano, M. Mapping Quantitative Trait Loci for Days-to-Heading, and Culm, Panicle and Internode Lengths in a BC1F3 Population Using an Elite Rice Variety, Koshihikari, as the Recurrent Parent. Breed. Sci. 2001, 51, 63–71. [Google Scholar] [CrossRef]
- Nagai, K.; Kuroha, T.; Ayano, M.; Kurokawa, Y.; Angeles-Shim, R.B.; Shim, J.-H.; Yasui, H.; Yoshimura, A.; Ashikari, M. Two Novel QTLs Regulate Internode Elongation in Deepwater Rice during the Early Vegetative Stage. Breed. Sci. 2012, 62, 178–185. [Google Scholar] [CrossRef]
- Bai, J.; Zhu, X.; Wang, Q.; Zhang, J.; Chen, H.; Dong, G.; Zhu, L.; Zheng, H.; Xie, Q.; Nian, J.; et al. Rice TUTOU1 Encodes a Suppressor of cAMP Receptor-like Protein That Is Important for Actin Organization and Panicle Development. Plant Physiol. 2015, 169, 1179–1191. [Google Scholar] [CrossRef]
- Nagai, K.; Kondo, Y.; Kitaoka, T.; Noda, T.; Kuroha, T.; Angeles-Shim, R.B.; Yasui, H.; Yoshimura, A.; Ashikari, M. QTL Analysis of Internode Elongation in Response to Gibberellin in Deepwater Rice. AoB Plants 2014, 6, plu028. [Google Scholar] [CrossRef]
- Qiao, B.-J.; Wang, Y.-Y.; Zhu, X.-B.; Hong, D.-L. QTL analysis of the uppermost internode length in rice under different growing environments. Yi Chuan 2007, 29, 1001–1007. [Google Scholar] [CrossRef]
- Hattori, Y.; Nagai, K.; Mori, H.; Kitano, H.; Matsuoka, M.; Ashikari, M. Mapping of Three QTLs That Regulate Internode Elongation in Deepwater Rice. Breed. Sci. 2008, 58, 39–46. [Google Scholar] [CrossRef]
- Park, J.-R.; Jang, Y.-H.; Kim, E.-G.; Hur, S.-S.; Kim, K.-M. Quantitative Trait Loci Mapping Identified Candidate Genes Involved in Plant Height Regulation in Rice. Int. J. Mol. Sci. 2023, 24, 16895. [Google Scholar] [CrossRef]
- Yue, B.; Xue, W.-Y.; Luo, L.-J.; Xing, Y.-Z. QTL Analysis for Flag Leaf Characteristics and Their Relationships with Yield and Yield Traits in Rice. Acta Genet. Sin. 2006, 33, 824–832. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Year | Mean ± SD | Min | Max | Skewness | Kurtosis | CV (%) | h2 |
|---|---|---|---|---|---|---|---|---|
| PH (cm) | 2021 | 111.23 ± 22.93 | 62.22 | 175.5 | 0.22 | −0.89 | 20.62 | 0.99 |
| 2022 | 112.20 ± 23.13 | 62.35 | 175 | 0.25 | −0.89 | 21.79 | 0.99 | |
| SD (cm) | 2021 | 7.4 1 ± 1.28 | 3.69 | 13.1 | 0.44 | 0.84 | 17.29 | 0.80 |
| 2022 | 7.40 ± 1.29 | 3.69 | 12.9 | 0.41 | 0.82 | 15.38 | 0.80 | |
| AT/S (Kpa) | 2021 | 9.06 ± 2.47 | 3.42 | 17.79 | 0.57 | 0.04 | 28.34 | 0.80 |
| 2022 | 9.05 ± 2.48 | 3.42 | 17.79 | 0.56 | 0.03 | 27.44 | 0.80 | |
| FirINL (cm) | 2021 | 34.86 ± 7.93 | 12.43 | 95.67 | 1.59 | 8.87 | 22.75 | 0.65 |
| 2022 | 34.85 ± 7.93 | 12.4 | 95.66 | 1.59 | 8.87 | 20.95 | 0.64 | |
| SedINL (cm) | 2021 | 23.46 ± 5.24 | 10.18 | 40.38 | 0.47 | 0.06 | 21.35 | 0.92 |
| 2022 | 23.58 ± 5.22 | 10.32 | 38.06 | 0.43 | −0.4 | 22.16 | 0.97 | |
| ThirINL (cm) | 2021 | 19.31 ± 5.45 | 6.82 | 32.93 | 0.29 | −0.87 | 28.35 | 0.94 |
| 2022 | 19 ± 5.48 | 6.5 | 32.61 | 0.27 | −0.87 | 28.85 | 0.94 | |
| ForINL (cm) | 2021 | 14.1 ± 6.05 | 1.96 | 31.44 | 0.42 | −0.75 | 42.91 | 0.93 |
| 2022 | 14.49 ± 6.05 | 2.43 | 31.73 | 0.41 | −0.77 | 41.73 | 0.92 | |
| FifINL (cm) | 2021 | 8.42 ± 5.42 | 0.94 | 28.88 | 0.59 | −0.66 | 45.35 | 0.92 |
| 2022 | 9.13 ± 5.44 | 0.71 | 29.58 | 0.57 | −0.67 | 46.63 | 0.92 |
| Source | df | SS | MS | Est. Var. | PMV% | p-Value |
|---|---|---|---|---|---|---|
| Among Pops | 2 | 13,681.113 | 6840.557 | 19.919 | 19% | p < 0.01 |
| Within Pops | 466 | 87,608.867 | 85.140 | 85.140 | 81% | p < 0.01 |
| Total | 468 | 101,289.981 | 105.058 | 100% |
| Subpopulation | Pop1 | Pop2 | Pop3 |
|---|---|---|---|
| Pop1 | 0.52 | 0.69 | |
| Pop2 | 0.56 | 0.58 | |
| Pop3 | 0.48 | 0.44 |
| Cluster | No. of LD a | Ratio b | Frequency of D′ c Value (p < 0.05) | Means of D′ | ||||
|---|---|---|---|---|---|---|---|---|
| Locus Pairs | (%) | 0–0.2 | 0.2–0.4 | 0.4–0.6 | 0.6–0.8 | 0.8–1.0 | ||
| POP1 | 1240 | 2.7 | 160 | 250 | 271 | 370 | 302 | 0.64 |
| POP2 | 725 | 4.7 | 96 | 266 | 265 | 145 | 193 | 0.61 |
| POP3 | 1437 | 2.4 | 49 | 227 | 361 | 335 | 190 | 0.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sowadan, O.; Xu, S.; Li, Y.; Muleke, E.M.; Sitoe, H.M.; Dang, X.; Jiang, J.; Dong, H.; Hong, D. Genome-Wide Association Analysis Unravels New Quantitative Trait Loci (QTLs) for Eight Lodging Resistance Constituent Traits in Rice (Oryza sativa L.). Genes 2024, 15, 105. https://doi.org/10.3390/genes15010105
Sowadan O, Xu S, Li Y, Muleke EM, Sitoe HM, Dang X, Jiang J, Dong H, Hong D. Genome-Wide Association Analysis Unravels New Quantitative Trait Loci (QTLs) for Eight Lodging Resistance Constituent Traits in Rice (Oryza sativa L.). Genes. 2024; 15(1):105. https://doi.org/10.3390/genes15010105
Chicago/Turabian StyleSowadan, Ognigamal, Shanbin Xu, Yulong Li, Everlyne Mmbone Muleke, Hélder Manuel Sitoe, Xiaojing Dang, Jianhua Jiang, Hui Dong, and Delin Hong. 2024. "Genome-Wide Association Analysis Unravels New Quantitative Trait Loci (QTLs) for Eight Lodging Resistance Constituent Traits in Rice (Oryza sativa L.)" Genes 15, no. 1: 105. https://doi.org/10.3390/genes15010105
APA StyleSowadan, O., Xu, S., Li, Y., Muleke, E. M., Sitoe, H. M., Dang, X., Jiang, J., Dong, H., & Hong, D. (2024). Genome-Wide Association Analysis Unravels New Quantitative Trait Loci (QTLs) for Eight Lodging Resistance Constituent Traits in Rice (Oryza sativa L.). Genes, 15(1), 105. https://doi.org/10.3390/genes15010105